


La mielofibrosis primaria es un padecimiento de la médula ósea caracterizado por fibrosis, asociado a esplenomegalia y hematopoyesis extramedular. En sangre periférica se observa un patrón de leucoeritroblastosis y dacriocitos, así como niveles elevados de citocinas inflamatorias y proangiogénicas.
Existen dos tipos: la mielofibrosis primaria, la cual no se asocia a ningún padecimiento medular, y la secundaria, que se asocia a patologías del síndrome mieloproliferativo como tombocitosis esencial o policitemia vera.
La incidencia en Estados Unidos es de 0.21-0.25 casos por cada 100 mil habitantes, se asocia con mayor frecuencia al sexo masculino y la media de edad al diagnóstico es de 67 años.
El tratamiento de la mielofibrosis primaria puede llevarse a cabo con diferentes modalidades, dependiendo del objetivo de tratamiento, ya sea con finalidad curativa o paliativa. Dentro de las modalidades se encuentran la cirugía con esplenectomía, el manejo farmacológico a base de medicamentos para producir inhibición celular como hidroxicarbamina, inmunomoduladores como talidomida, o terapia dirigida como ruxolitinib. La radioterapia generalmente es empleada para tratar la esplenomegalia refractaria al tratamiento médico o en aquellos pacientes con alguna contraindicación quirúrgica y, finalmente, el trasplante de médula ósea ha mostrado ser el único tratamiento que modifica la supervivencia.
Primary myelofibrosis is a disease of the bone marrow characterized by fibrosis, associated with splenomegaly and extramedullary haematopoiesis. In peripheral blood, a pattern of leucoerythroblastosis and tear drop cells, as well as elevated levels of pro-inflammatory and angiogenic cytokines is observed.
There are two types: primary myelofibrosis, which is not associated with any bone marrow condition, and secondary disease, which is associated with myeloproliferative syndrome conditions such as essential thrombocytosis or polycythaemia vera.
The incidence in the United States ranges from 0.21 to 0.25 cases per 100 thousand inhabitants, it is associated more often with the male gender, and mean age at diagnosis is 67 years.
The treatment of primary myelofibrosis can be in different forms, depending on the goal of treatment, with curative or palliative intent. Treatment modalities include surgical procedures with splenectomy, pharmacological treatment with drugs to produce cell inhibition such as hydroxycarbamide, immunomodulators such as thalidomide, or targeted therapy such as ruxolitinib; radiation therapy is generally used to treat medical treatment-refractory splenomegaly or in those patients with any surgical contraindication and, finally, bone marrow transplantation has proven to be the only treatment that modifies survival.
La mielofibrosis es una patología que fue descrita por primera vez por el cirujano alemán Gustav Heuck (1854-1940) en 18791, en tanto que Silverstein definió los conceptos establecidos en la primera mitad del siglo XX para explicar la patogenia de esta enfermedad, su origen en la médula ósea, la hematopoyesis extramedular y la relación de la fibrosis con los cambios hematopoyéticos.
Esta patología se caracteriza por afección de la médula ósea y forma parte de los síndromes mieloproliferativos. Los sinónimos de esta entidad son mielofibrosis crónica idiopática, mielofibrosis con metaplasia mieloide agnogénica, osteomieloesclerosis y mieloesclerosis2.
Una de sus características distintivas radica en la proliferación clonal de células mieloides con megacariocitopoyesis. Esta población celular anormal libera citocinas y factores de crecimiento como el factor de crecimiento derivado de plaquetas (platelet-derived growth factor), el factor de crecimiento trasformador beta (transforming growth factor beta), el factor de crecimiento del endotelio vascular (vascular endothelium growth factor) y calmodulina, con la producción de fibrosis como efecto secundario y la infiltración de órganos como hígado y bazo2. En el 50% de los casos se asocia la mutación del gen JAK2 (Janus cinasa mutado 2)2.
Otras características incluyen anemia, fibrosis de la médula, esplenomegalia progresiva, osteoesclerosis y aumento de células CD34 +; en el frotis de sangre periférica se observan eritrocitos en forma de lágrima o dacriocitos, elementos mieoloides y eritroides inmaduros (patrón de leucoeritroblastosis). La supervivencia en promedio es de 5 años; sin embargo la calidad de vida se ve fuertemente afectada.
Caso clínicoPaciente de 58 años de edad, originaria y residente del Estado de México, sin antecedentes de importancia.
Inició su padecimiento en el año 2010 con dolor abdominal localizado en el cuadrante inferior izquierdo, asociado a saciedad temprana y tumoración palpable, con esplenomegalia de 10cm debajo del borde costal, por lo que inicialmente se diagnosticó leucemia mieloide, manejada con imatinib en ese mismo año.
Sin embargo al no mostrar mejoría y presentar efectos adversos del medicamento como edema, parestesias y palpitaciones, la paciente suspendió el tratamiento.
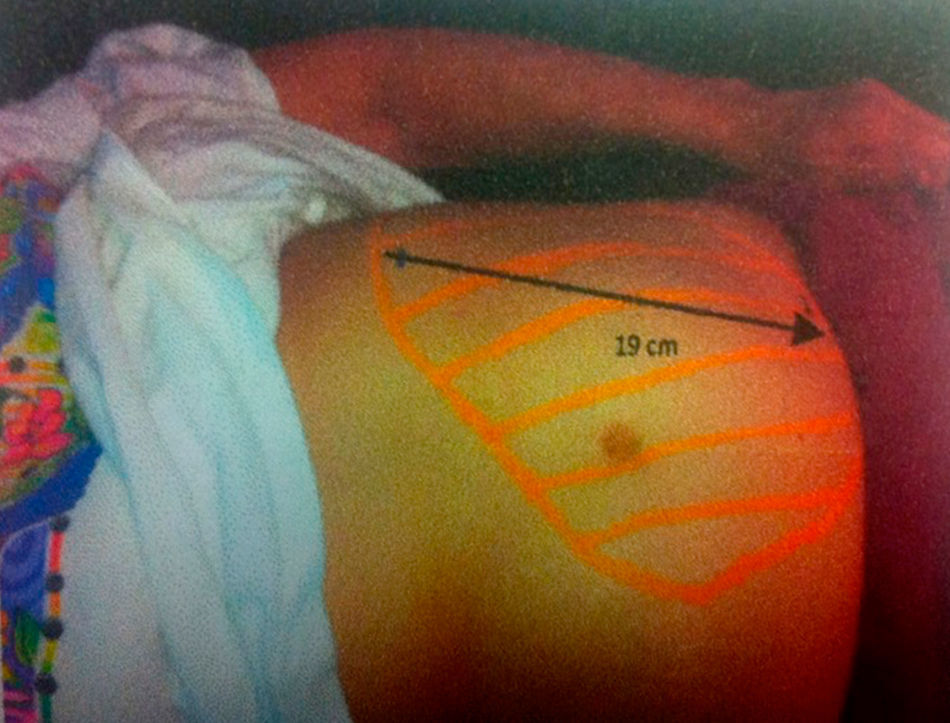
Tres años después, se detectó un aumento del tamaño del bazo de 19cm por debajo del borde costal (fig. 1), motivo por el cual se protocolizó nuevamente para diagnóstico, encontrándose positividad a la mutación del gen JAK 2, negatividad al gen quimérico BCR/ABL y fibrosis grado III en la biopsia de médula ósea.

Por lo tanto, el diagnóstico se estableció como mielofibrosis primaria y se inició el tratamiento con ruxolitinib 25mg cada 12 h, respondiendo parcialmente con una disminución del tamaño del bazo (fig. 2).

Sin embargo, a pesar del manejo, continuó con progresión de la enfermedad, caracterizada por la aparición de adenopatías en el cuello, las cuales en un inicio midieron 3cm y eran de características blandas, móviles, indoloras. Al cabo de 3 meses de evolución, la paciente fue enviada a la Unidad de Radioterapia por parte de Hematología para el tratamiento de las adenopatías cervicales.
En ese momento, la paciente se encontraba con limitación en la movilidad de cuello, con adenomegalias pétreas, fijas, de 10 x 8cm en el hemicuello izquierdo, las cuales abarcaban los niveles ganglionares IB-IV. En el hemicuello derecho, presentaba un conglomerado ganglionar de 3 x 5cm con afección del nivel II.
A la palpación de la región axilar, se encontró presencia de un conglomerado ganglionar de 5cm, de consistencia ahulada, móvil, indolora y en la exploración abdominal se palpó esplenomegalia con 22cm por debajo del borde costal.
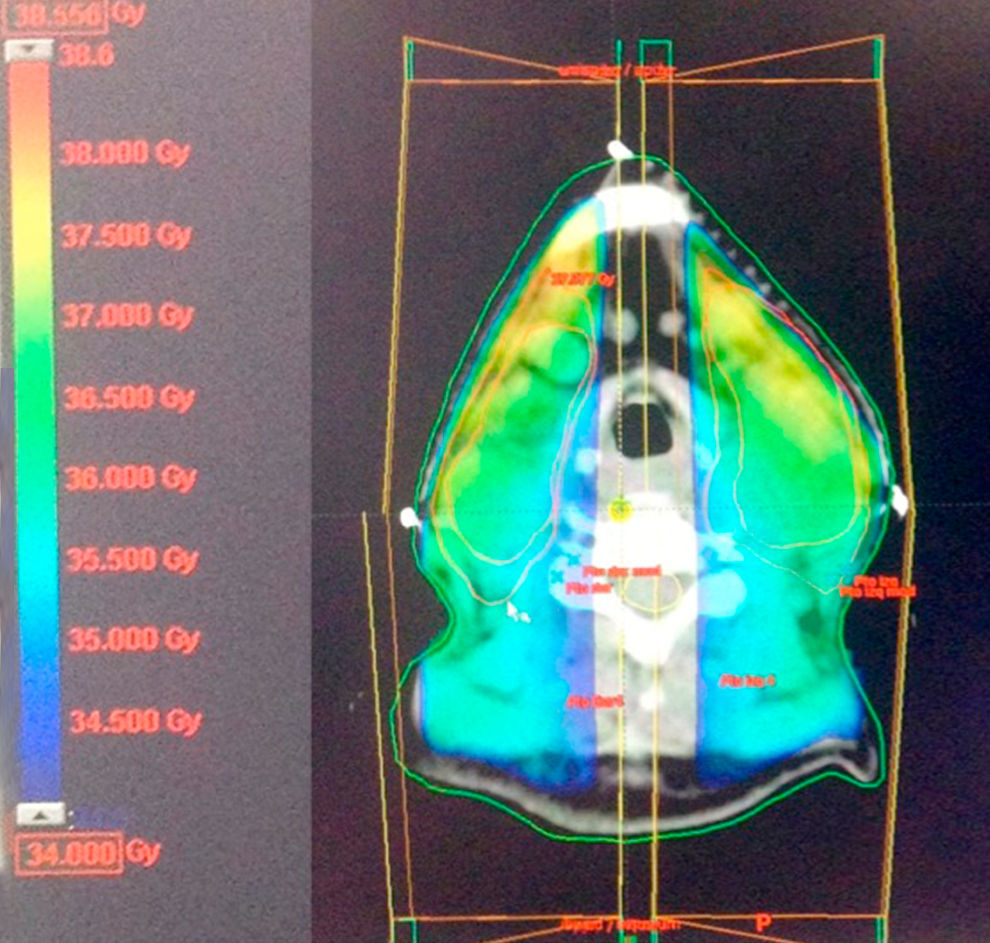
Fue considerada candidata a radioterapia para la afección ganglionar cervical con una dosis de 34Gy en 17 sesiones, con un fraccionamiento convencional de 2 Gy por fracción, con técnica conformal (3D), utilizando dos campos opuestos paralelos (fig. 3). Durante el tratamiento no se presentó toxicidad gastrointestinal o dermatológica.
 con 2 campos opuestos paralelos, con prescripción de 34Gy, con dosis de 2Gy por sesión.")
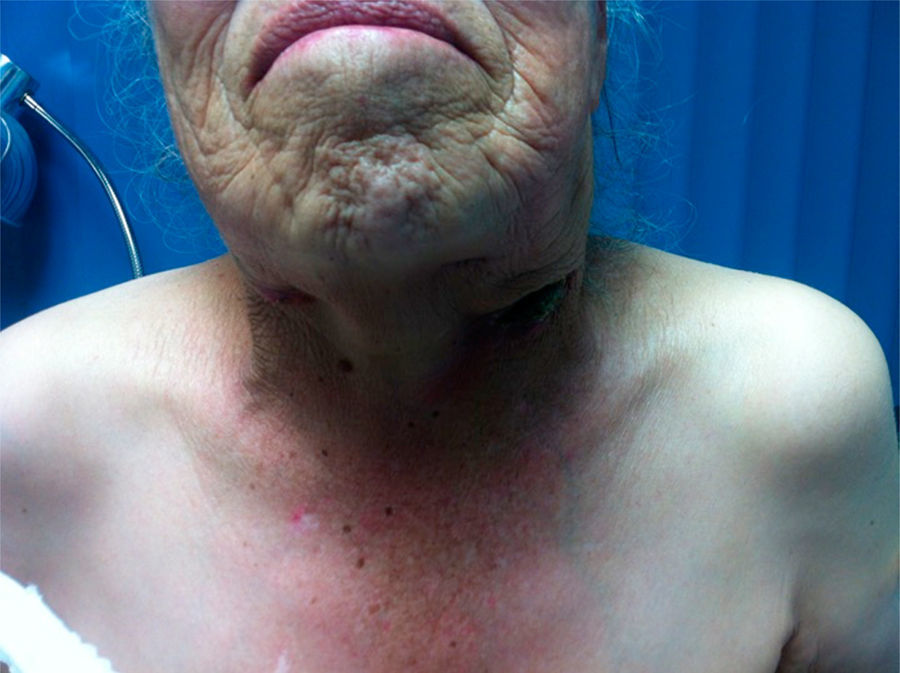
Tras la radioterapia, la respuesta ganglionar fue parcial con adenomegalias que se tornaron móviles y con reducción de volumen en el hemicuello izquierdo a 1.5 x 1.5cm y en el hemicuello derecho a 4 x 5cm (fig. 4).
Discusión
La mielofibrosis (MF) es una patología poco frecuente3. En las estadísticas del Programa de Vigilancia, Epidemiología y Resultados Finales (Surveillance, Epidemiology, and End Results Program), la tasa de incidencia es de 0.21; la mortalidad se encuentra asociada en más del 30% de los casos a leucemias, en el 10% a infecciones, en el 5% a sangrado e hipertensión portal y en menos del 5% a trombosis4. Se ven afectados en la mayoría de los casos pacientes mayores de 60 años5; sin embargo un 10% del padecimiento afecta a individuos < 45 años.
La etiología de la mielofibrosis incluye enfermedades autoinmunitarias (lupus eritematoso sistémico, esclerodermia, mielofibrosis autoinmune), agentes tóxicos (benceno, radiación ionizante), infecciones (tuberculosis, leishmaniasis, VIH), neoplasias (linfomas, leucemia eosinofílica crónica, enfermedades mieloproliferativas clásicas, mieloma múltiple), deficiencia de vitamina D, osteopetrosis, osteodistrofia renal, hiperparatiroidismo y enfermedad de Paget2.
Para su clasificación se toma en cuenta el consenso del Grupo de Trabajo Internacional para la Investigación y el Tratamiento de la Mielofibrosis (International Working Group for Myelofibrosis Research and Treatment) de noviembre de 20062:
MF primaria: aparición de novo de la enfermedad, no asociada a policitemia vera ni trombocitemia esencial.
MF pospolicitemia vera o postrombocitemia esencial.
Fase blástica de MF y pospolicitemia vera /PTR-MF; corresponde a la trasformación leucémica.
La supervivencia a 3 años es del 52%, y el tiempo promedio hasta la progresión de 7 meses6. En el 4-20% de los casos se presenta la trasformación a leucemia, normalmente dentro de los primeros 10 años posteriores al diagnóstico, y se ha documentado que se vincula a un peor pronóstico.
En el 45-68% de los casos se observa la presencia de mutación del gen JAK2 V617F1,7,8, importante para el diagnóstico y en caso de negatividad, se lleva a cabo la determinación del gen quimérico BCR/ABL1 y del reordenamiento de PDGFRA y PDGFRB, los cuales deben excluirse en presencia de eosinofilia. La mutación del oncogén TET2, presente el 15% de los casos, se asocia a la edad avanzada. La mutación MPL W515L se presenta en el 9% de los casos, asociada principalmente a pacientes femeninos y geriátricos, y se manifiesta con anemia severa8.
La enfermedad tiene dos fases: estadio prefibrótico y estadio de fibrosis manifiesta2.
El primer estadio se caracteriza por un incremento en la producción de granulocitos y megacariocitos displásicos, así como presencia de fibrosis de la médula ósea grado 1 (trama laxa de reticulina). Los pacientes generalmente se encuentran asintomáticos, pero existen alteraciones como anemia refractaria, leucocitosis, trombocitosis, e incluso esplenomegalia.
El segundo estadio se caracteriza por osteoesclerosis, megacariocitopoyesis intrasinusoidal, leucoeritroblastosis o abundancia de dacriocitos. La fibrosis en la médula ósea es grado 2-3 (grado 2: incremento difuso y denso de reticulina y osteoesclerosis focal; grado 3: incremento extenso y difuso de reticulina, con haces de colágeno, asociado a osteoesclerosis significativa)9. Asimismo, se observa anemia importante, esplenomegalia evidente y otros síntomas constitucionales.
El diagnóstico es clínico, citogénico y anatomopatológico2. Al inicio, la enfermedad es asintomática, con anemia y trombocitosis con o sin alteraciones morfológicas en los eritrocitos. Por su parte, la enfermedad avanzada se caracteriza por esplenomegalia asociada a saciedad precoz, tumoración palpable, dolor que puede ser secundario a infarto esplénico, distensión abdominal, datos de hipertensión portal, e hipertensión pulmonar y citopenias.
Un 10% de los individuos afectados experimenta síntomas B. La hematopoyesis extramedular es causante de organomegalia, caracterizada más frecuentemente por esplenomegalia y, en segundo lugar, por hepatomegalia3. A nivel óseo, los signos son consecuentes a osteoesclerosis y periostitis.
En sangre periférica se observa leucoeritroblastosis y presencia de dacriocitos3. Otros datos incluyen:
Elevación de deshidrogenasa láctica por hematopoyesis inefectiva, alteraciones de las pruebas funcionales hepáticas por infiltración hepática o cirrosis, alteración en las pruebas de coagulación por el compromiso hepático, además, puede existir coagulación intravascular diseminada en forma subclínica2.
Por su parte, el Comité europeo de Hematología recomienda determinar la presencia de mutación del gen JAK2 V617F y excluir mutaciones de BCR/ABL, PDGFRA y PDGFRB en caso de eosinofilia. La mutación de JAK2V617F es un factor que contribuye a la existencia de la enfermedad, más no es determinante de la misma2.
En cuanto a los criterios diagnósticos10, estos se clasifican en mayores y menores. Los criterios mayores son denominados A, donde A1 corresponde a fibrosis de la médula ósea y A2 define la mutación del gen JAK2, MPL o ausencia de BCR/ABL1. Por su parte los criterios B comprenden esplenomegalia idiopática palpable, patrón leucoeritroblástico, presencia de dacriocitos, síntomas constitucionales y evidencia de hematopoyesis ineficaz10. Los criterios A1 y A2 más 3 criterios clasificados como B, establecen el diagnóstico.
El pronóstico se determina con el Sistema Internacional de Puntaje Pronóstico (International Prognostic Scoring System [IPSS]), el cual clasifica el riesgo como bajo, intermedio 1, intermedio 2 y alto según el puntaje asignado. El IPSS otorga un punto por cada uno de los siguientes factores: edad > 65 años, síntomas constitucionales, hemoglobina < 10g/dl, leucocitos > 25 109/l y blastos >1%2,7,11,12.
La supervivencia con la clasificación de riesgo bajo con 0 puntos es de 11.3 años, en riesgo intermedio 1 con un punto es de 7.9 años, en riesgo intermedio 2 con 2 puntos de 4 años y en riesgo alto con > 3 puntos de 2.3 años3,13.
La supervivencia estimada es de 5 a 7 años y se ve comprometida por las transfusiones sanguíneas frecuentes, la esplenomegalia masiva y síntomas constitucionales13.
El tratamiento quirúrgico con esplenectomía está indicado cuando existe la presencia de caquexia, anemia, hipertensión portal o esplenomegalia sintomática refractaria a la medicación (nivel de evidencia 2, grado c).
El tratamiento médico se basa en el uso de hidroxicarbamina en ausencia de citopenias, con una tasa de respuesta del 45%; sin embargo no produce ningún beneficio de supervivencia14. Por otro lado, con fármacos inmunomoduladores como talidomida, lenalidomida y prednisona se ha documentado una respuesta global del 33%, con índices de trombocitopenia del 50%, anemia del 22% y esplenomegalia del 8%. Estos medicamentos pueden ser empleados en pacientes con citopenias. Finalmente, se ha sugerido el uso de inhibidores de JAk como parte de ensayos clínicos en pacientes con riesgo intermedio 2 o > 45 años de edad.
En el rubro de las indicaciones de radioterapia se encuentra el dolor óseo, pacientes no candidatos a esplenectomía y hematopoyesis extramedular con compromiso de órganos vitales3. La dosis no está bien definida; se han empleado dosis tan bajas como 2.77Gy en 7.5 sesiones con reducción del tamaño esplénico; sin embargo el 44% de los sujetos presentó citopenias. Un esquema adyuvante a la esplenectomía es radiación con dosis bajas de 1Gy cada uno a 3 meses; sin embargo, esta pauta también se asocia a pancitopenias.
En el caso que nos ocupa se decidió utilizar una dosis similar a la que se maneja para la actividad ganglionar por enfermedad linfoproliferativa, como en el caso de los linfomas. Se administraron 34Gy, lográndose una reducción importante del conglomerado ganglionar una vez terminado el tratamiento, con una adecuada tolerancia al mismo.
El tratamiento de la hepatomegalia con radioterapia se sugiere con dosis de 9Gy en 6 sesiones (1.5Gy/sesión), obteniéndose una disminución del tamaño del órgano en un 35% y hasta un 86% de los pacientes alcanza una respuesta transitoria por tres meses3. La paciente descrita en el caso clínico satisface los criterios para beneficiarse de irradiación esplénica, por el tamaño del bazo y al no ser una candidata quirúrgica, sin embargo, esta no se consideró debido a que la paciente se encontraba en tratamiento con terapia dirigida, en seguimiento por el Servicio de Hematología bajo protocolo de investigación.
Por otro lado, el trasplante de médula ósea está indicado en pacientes < 45 años, con IPSS intermedio 2 o alto y presenta un alto riesgo de morbimortalidad, con índices de supervivencia libre de enfermedad a 5 años del 33%, supervivencia global a 7 años del 61%, mortalidad a 5 años del 35% y tasas de recaída del 29%.
Finalmente, el seguimiento se basa en la determinación de los niveles de deshidrogenasa láctica, pruebas funcionales hepáticas mensuales, cuenta de CD34, ultrasonografía abdominal cada 6-12 meses y biopsia de médula ósea.
ConclusiónLos síndromes mieloproliferativos son de naturaleza heterogénea y difíciles de tratar; si bien las opciones terapéuticas siguen siendo limitadas, la radioterapia representa una opción en casos refractarios a manejo médico o no candidatos a cirugía, y se recomienda en todo caso, la conducta terapéutica deberá individualizarse en cada paciente.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.