


¿ INTRODUCCIÓN
Los sarcomas son tumores malignos que se originan de tejidos blandos, hueso, piel u órganos internos. Los originados en partes blandas son en su gran mayoría benignos y 100 veces más comunes que los malignos. Debido a que hay más de 50 subtipos histológicos de sarcomas de partes blandas, su precisión diagnostica es difícil.1 En 2008 se documentó que 10 000 americanos fueron diagnosticados con esta neoplasia, de los cuales 3500 morirán por esta causa.2 Este tipo de tumores se localiza en la profundidad de los tejidos blandos de extremidades inferiores, sobre todo el muslo. Se han visto en otras áreas como tórax, hombro, ingle y raramente interior del abdomen.3 El sarcoma fibromixoide de bajo grado es una neoplasia de tejidos blandos recientemente descubierta y poco común. Fue reportado originalmente por Evans en 1987, quien describió 10 casos adicionales en 1993. Desde entonces algunos casos esporádicos han sido reportados. Anteriormente se creía que era una variante del histiocitoma fibroso maligno.4 Tiene un comportamiento agresivo con una tasa alta de recurrencia local (33%) y metástasis (58%). Su incidencia es incierta. Se han documentado alrededor de 150 casos a nivel mundial con predominio en el sexo masculino y adultos jóvenes del grupo comprendido entre 25 y 46 años, aunque se ha presentado en sujetos mayores (56 años).5
Debido a la incidencia poco común de esta neoplasia, se describe un caso de localización en la región lumbosacra. Asimismo se realiza una revisión de la literatura internacional para conocer las características clínicas, histopatológicas y diagnósticas, así como las opciones terapéuticas existentes.
El objetivo de este artículo es describir un caso de sarcoma variedad fibromixoide de bajo grado presentado en un paciente masculino en una localización inusual, además de detallar las características clínico-patológicas y el manejo integral de esta neoplasia. Su incidencia es baja en nuestro país y en el mundo. Se realiza también una revisión exhaustiva de la literatura sobre el tema.
¿ PRESENTACIÓN DEL CASO
Paciente masculino de 36 años edad, sin antecedentes patológicos de importancia. Refiere iniciar hace 20 años con aumento de volumen en región lumbosacra derecha, asintomático hasta hace tres años cuando se agrega dolor en el área; el dolor aumentaba con la movilización activa, sin mejoría a la administración de analgésicos hasta hacerse incapacitante.
Posteriormente se acentúa el dolor en marzo de 2010, motivo de hospitalización. Se inicia protocolo de estudio, tomando tomografía axial computarizada (TAC) de región lumbosacra donde se observa tumoración lumbar derecha de 10 x 15 cm, sin aparente infiltración a tejido óseo. El paciente es intervenido quirúrgicamente fuera de nuestra unidad, realizándose resección amplia de tumoración el 30 de marzo de 2010. Se reportan hallazgos de tumor con extensión a cresta ilíaca y columna lumbar, con residual macroscópico y reporte histopatológico de sarcoma fibromixoide. Ante esto es enviado a nuestra unidad para valoración de tratamiento adyuvante con radioterapia.
El paciente es visto por primera vez en el servicio de oncología quirúrgica el 30 de mayo de 2010. Durante la exploración física se observa sobre línea media cicatriz de 16 cm de longitud de la cirugía previa con datos de actividad tumoral de aproximadamente 3 cm, además de tumor en región lumbosacra bilateral de 4 cm, sin adenopatías regionales; resto de la exploración sin datos patológicos. La revisión de laminillas el 18 de junio de 2010 en nuestra unidad reporta sarcoma fibromixoide de bajo grado con diferenciación a músculo liso. La inmunohistoquímica reporta positividad para vimentina y proteína S-100, CD68 positivo en el 10% de las células neoplásicas y actina de músculo liso focalmente positiva en el área hipocelular. El 28 de junio de 2010 se realiza estudio ultrasonográfico (USG) hepático que se reporta sin datos de actividad tumoral en hígado. Se toma una telerradiografía de tórax sin que se observen datos de actividad tumoral metastásica.
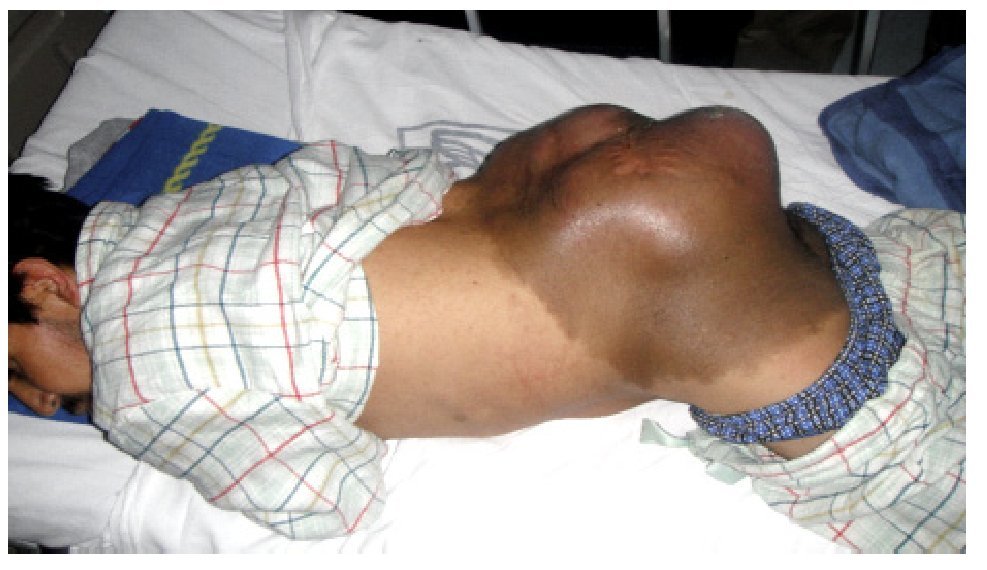
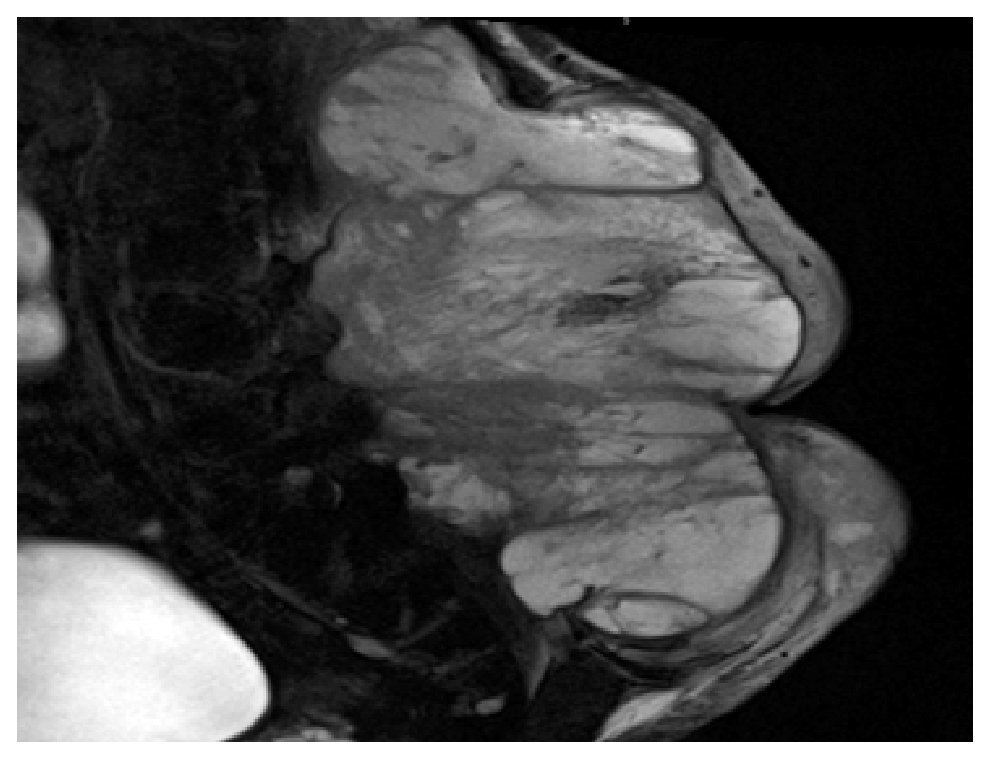
El paciente es valorado en el servicio de radioterapia el 28 de junio de 2010, programándose inicio de radioterapia (dosis total 66 Gy) sin que acuda a la cita. Visto posteriormente en la consulta externa de oncología quirúrgica (11 de noviembre de 2010), refiere dolor en sitio de tumoración así como alteraciones sensitivas y motoras en extremidades inferiores. Durante la exploración se detecta tumor en región lumbosacra bilateral de 35 x 40 cm, lobulado, exofítico, coloración café-violácea, fijo a columna lumbar (Figura 1). Es hospitalizado para protocolo de preparación quirúrgica; el 17 de noviembre de 2010 se realiza resonancia magnética nuclear (RMN) que evidencia lesión prevertebral de tejidos blandos, la cual se extiende desde L1 hasta región sacra y mide 32 cm rostro caudal y 33 cm ventrodorsal sin invadir conducto medular (Figura 2). Es valorado por el servicio de neurocirugía descartándose daño medular.
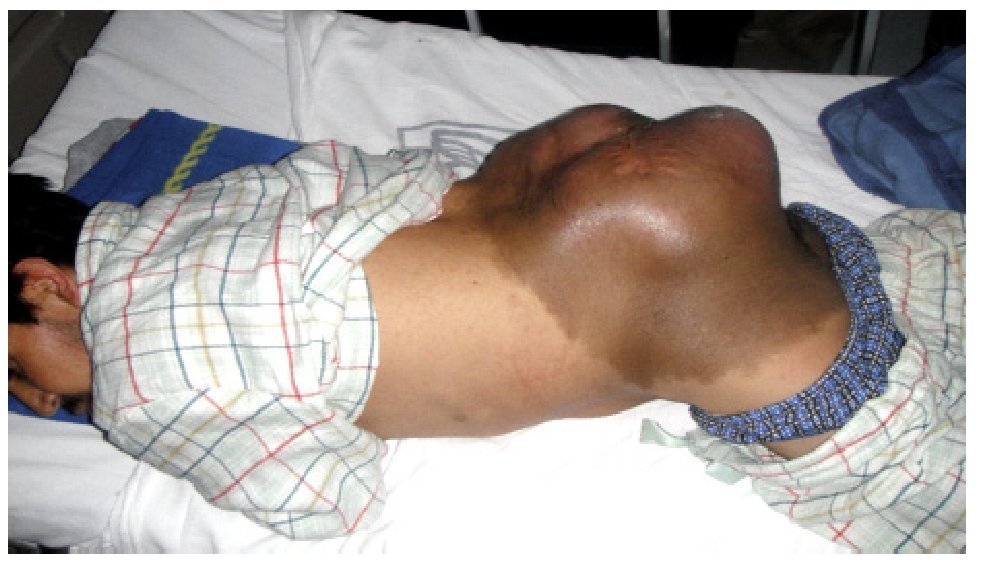
Figura 1. Tumor de 35 x 40 cm en región lumbosacra, lobulado, exofitico, coloración caféviolácea, fijo a columna lumbar.
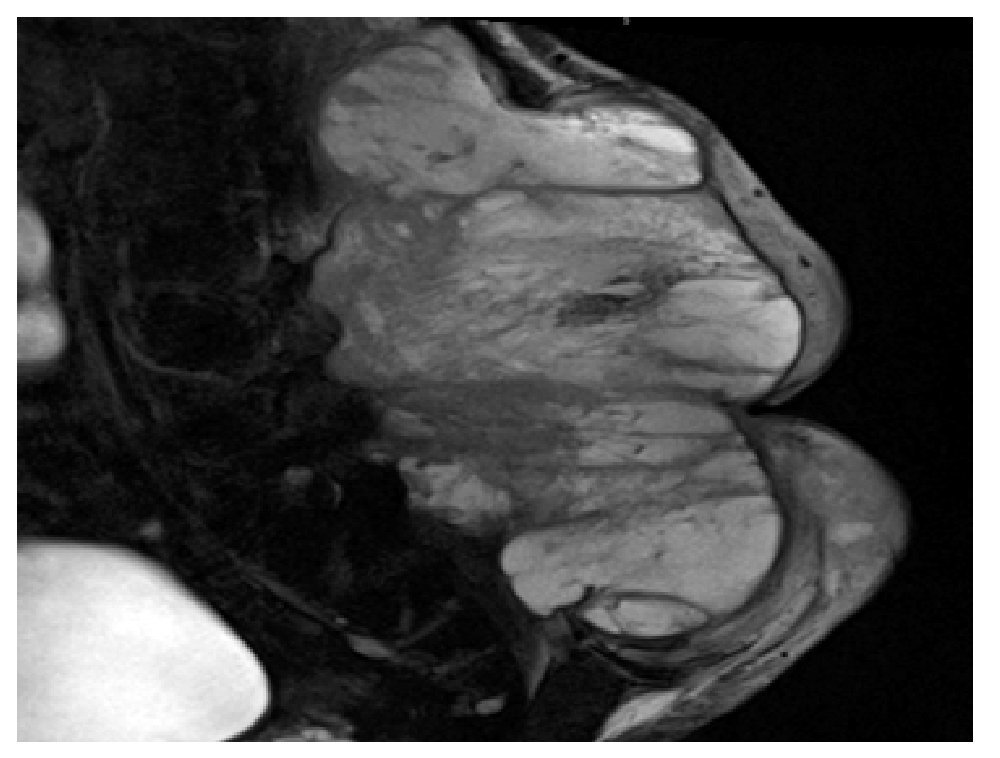
Figura 2. Imagen de resonancia magnética en T2 donde se observa lesión heterogénea con realce y reforzamiento periférico de tejidos blandos, prevertebral que mide 32 cm rostro caudal y 33 cm ventrodorsal.
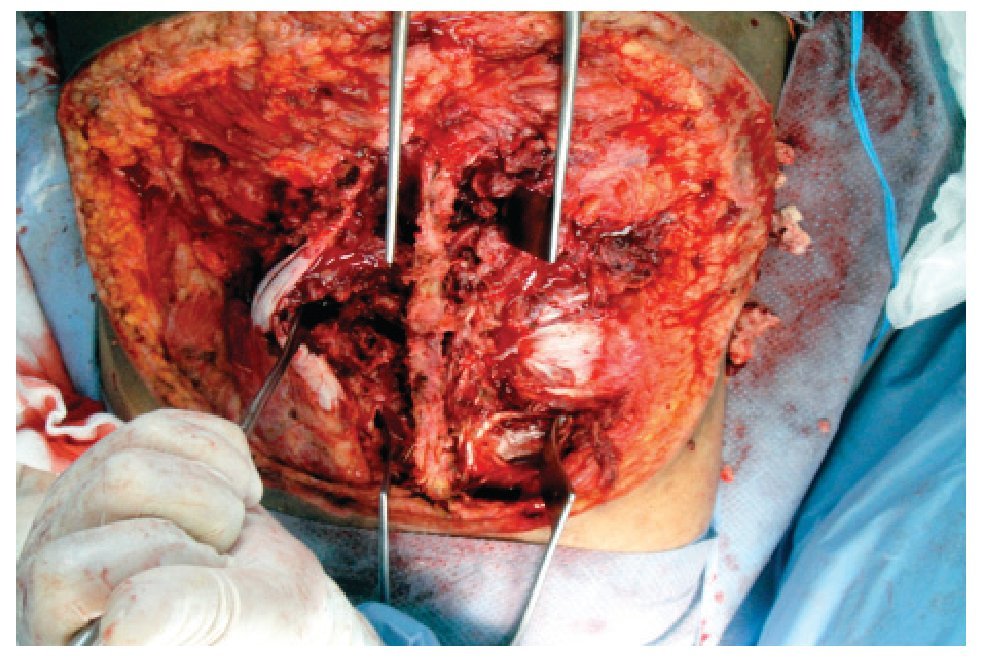
El 19 de noviembre de 2010 se interviene quirúrgicamente en conjunto con el servicio de neurocirugía y cirugía plástica y reconstructiva, realizándose resección amplia de la tumoración, denervación facetaria y rotación de colgajos con cobertura del 50% del defecto y colocación de sistema VAC. Se reportan como hallazgos áreas de necrosis y hemorragia dentro de la tumoración con afección de tejidos blandos de columna lumbosacra desde T12 hasta la punta de sacro, sin invasión de canal medular (Figura 3).
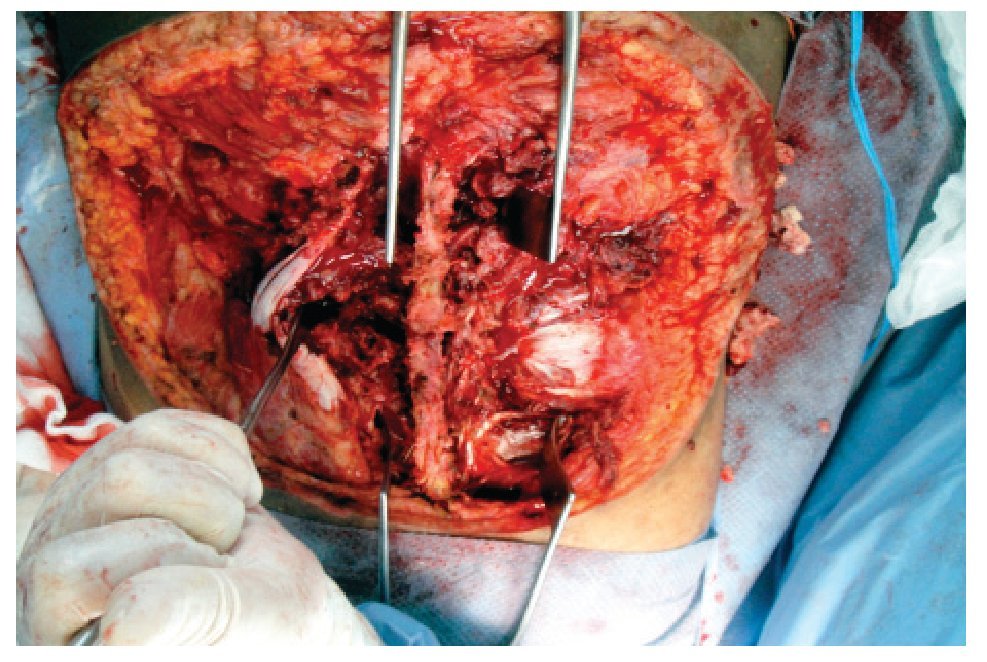
Figura 3. Imagen correspondiente a la resección amplia de la tumoración con denervación facetaria; con áreas de necrosis y hemorragia dentro de la tumoración y afección de tejidos blandos de columna lumbosacra.
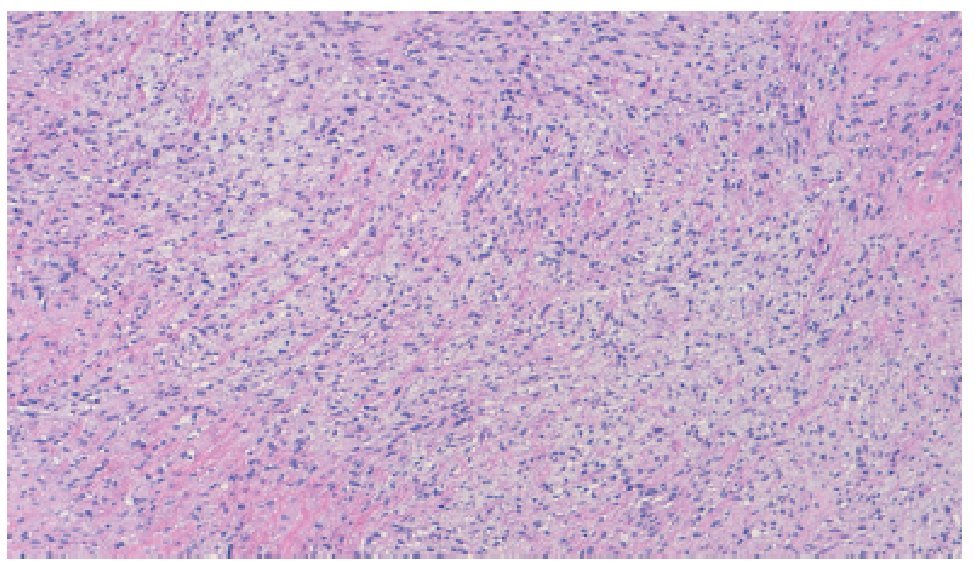
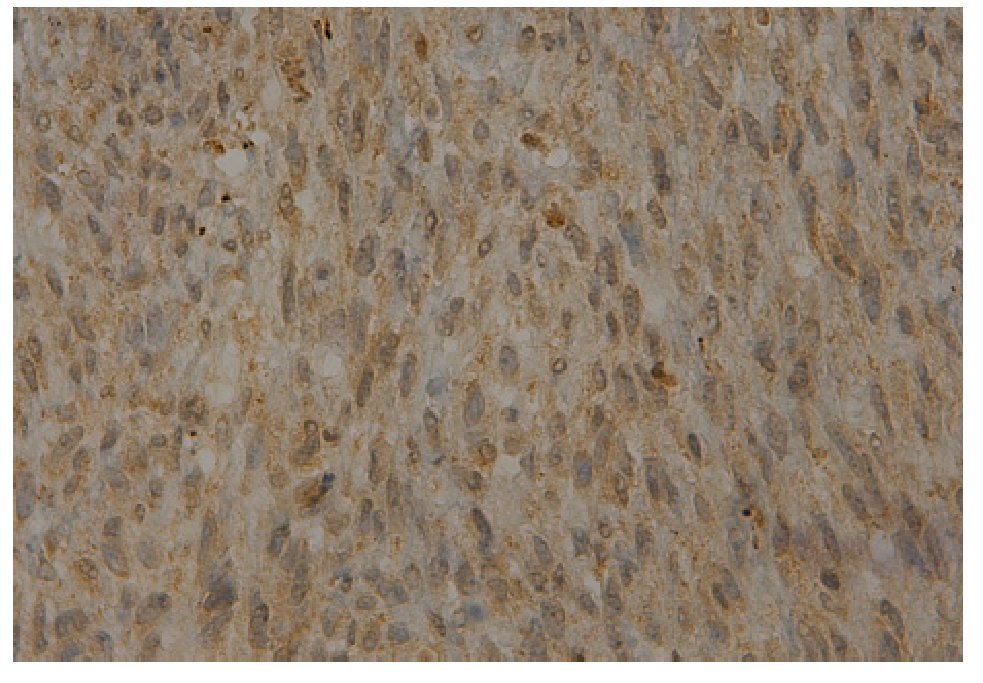
El reporte histopatológico de la pieza informa sarcoma fibromixoide (Figura 4) con bordes quirúrgicos infiltrados por tumor. En la inmunohistoquímica se reporta positividad a vimentina y actina muscular específica (Figura 5).
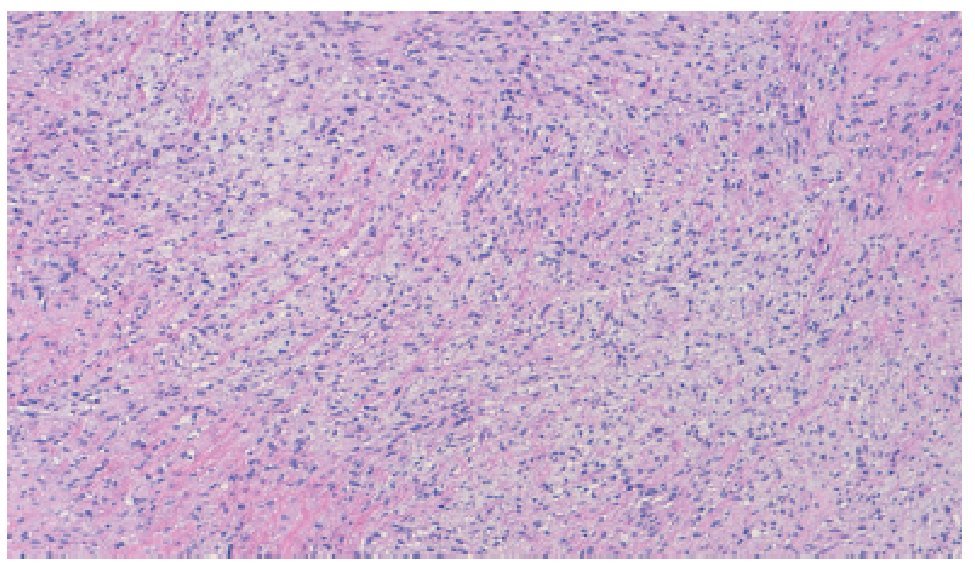
Figura 4. Microfotografía donde se observa el patrón con matriz fibromixoide con áreas de fibrosis densa (tinción H y E 10X).
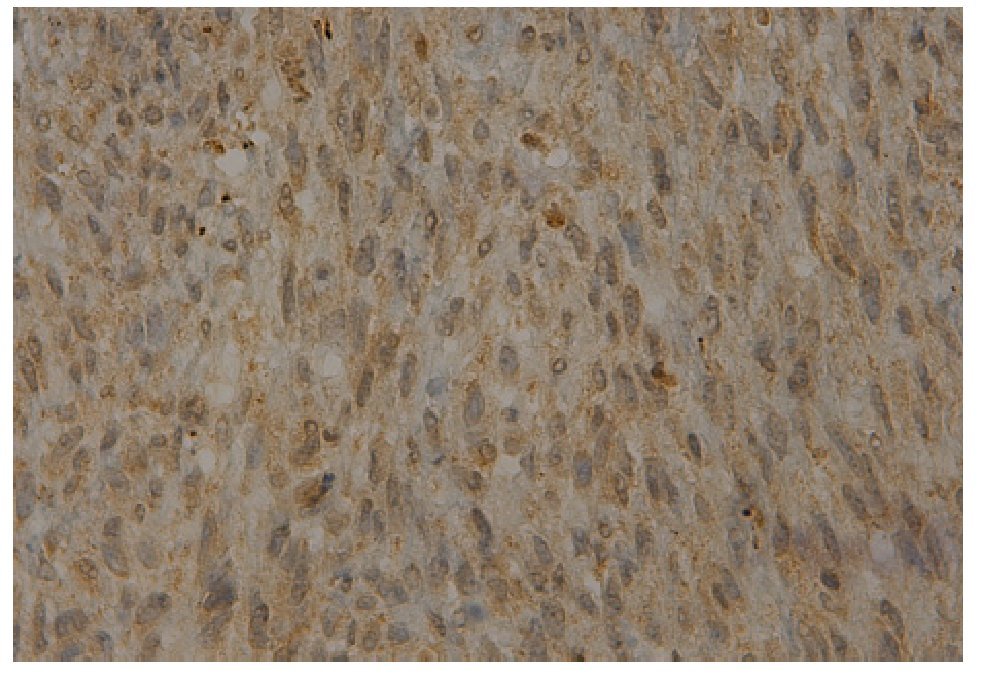
Figura 5. Inmunorreacción a la actina muscular específica.
El 30 de noviembre de 2010 se realiza lavado quirúrgico y debridación por necrosis del colgajo. Se continúa vigilancia en la consulta externa de oncología quirúrgica. El paciente refiere astenia y adinamia; en la exploración física se observa herida cruenta con exposición de columna lumbosacra, así como datos de actividad tumoral y zonas de necrosis. Es valorado por el servicio de radioterapia el 21 de enero de 2011, donde se propone radioterapia paliativa única de 6 Gy a hemicuerpo. Es hospitalizado en marzo de 2011 por mal estado general, desequilibrio hidroelectrolitico, síndrome anémico y datos de insuficiencia renal aguda. Se mantiene sólo con medidas paliativas por estar fuera de tratamiento oncológico y se estabiliza durante su estancia hasta el 15 de marzo de 2011 cuando es dado de alta voluntaria a su domicilio.
¿ DISCUSIÓN
El sarcoma fibromixoide es una variante del fibrosarcoma; se trata de un tumor raro, con un alto potencial de metástasis a pesar de su apariencia histológica benigna. Algunas veces el largo intervalo entre su presentación y la metástasis causa problemas al patólogo, radiólogo y cirujano. El tumor se presenta usualmente en las extremidades inferiores (50%) sobre todo en muslo, también en tronco, región inguinal y hombro. Esporádicamente puede encontrarse en localizaciones como retroperitoneo, cabeza (2.7%), axila, hombro, tórax o región paravertebral.6 La mayoría se localiza de forma subfascial; raramente tiene localización subcutánea o dérmica. Se presenta en adultos jóvenes y de edad media. También se ha visto en edad pediátrica aunque esta histología es muy rara en estos pacientes.7-9
Dentro de las alteraciones genéticas encontradas en esta patología, a través de pruebas con RT-PCR (reacción en cadena de polimerasa por transcriptasa inversa), FISH (hibridación in situ fluorescente), reacción en cadena de polimerasa por ADN y bloques de parafina, se observa la translocación t (7; 16), (q34; p11) y fusión entre los genes FUS y CREB3L2 en el 95% de los casos, y en un 5% la variante FUS-CREB3L1 confirmada por múltiples estudios.10-12
Histológicamente se caracteriza por presentar una matriz de colágeno con zonas de hipocelularidad y muchos nódulos celulares mixoides. Las células tumorales son usualmente pequeñas con escaso citoplasma eosinofílico, núcleo de redondo a ovoide y ausencia de nucléolo. No obstante hay zonas focales atípicas de hipercelularidad, actividad mitótica aumentada, hipercromatismo nuclear y necrosis que pueden verse en el 10% de los casos. La coloración con inmunohistoquímica es positiva a vimentina y negativa a desmina, queratina, proteína S-100, CD34 y CD31. El antígeno de membrana epitelial se ha reportado positivo en algunos casos.12,13
La actina muscular específica es positiva en la pared de vasos pequeños dentro del tumor y fuertemente positiva en la capa fibrosa periférica. Existe una entidad denominada tumor de células hialinizantes en forma de rosetas gigantes que comparte ciertas características histológicas y genéticas con el sarcoma fibromixoide.12-14
La presentación clínica suele ser de larga evolución, con un tumor de tejidos blandos de lento crecimiento, poco doloroso y confinado al sitio afectado. Es raro que se presente como una masa dolorosa; en ocasiones cursa con hemorragia tumoral y es infrecuente que origine síntomas neurológicos o vasculares.15,16 De presentarse la tumoración en la pared torácica los pacientes pueden experimentar de forma aguda distrés respiratorio y dolor torácico.6 En cuanto al diagnóstico diferencial, éste incluye lesiones que muestran una proliferación celular con patrón mixoide con o sin componente fibroso. Las entidades con patrón mixoide predominante sin componente fibroso significativo incluyen mixoma, mixofibrosarcoma de bajo grado, angiomixoma, liposarcoma mixoide y neurofibroma mixoide. Los tumores con morfología mixoide y fibrosa incluyen al neurofibroma, fibromatosis, perineurioma e histiocitoma fibroso. Otras entidades abarcan al tumor desmoide, fibrosarcoma desmoplásico y liposarcoma desdiferenciado de bajo grado.17,18
El diagnóstico se realiza mediante biopsia de toda lesión tumoral que persista más de cuatro semanas, sea sintomática o mayor de 5 cm. Hay diferentes modalidades: a) biopsia incisional: se realiza en tumores de más de 5 cm, la incisión debe efectuarse en el sitio más prominente, paralela al grupo muscular o longitudinal a la extremidad; se deben evitar los drenajes (si no es posible se deben exteriorizar a través de la herida principal) con la finalidad de que al momento de la resección quirúrgica ésta sea incluida en el espécimen de la pieza; b) biopsia excisional: se realiza en tumores de 3 a 5 cm, superficiales y en sitios con posibilidad de realizar un cierre primario de la herida; c) biopsia por aspiración con aguja fina (BAAF): sólo para diagnóstico de recurrencia o metástasis tumoral (sensibilidad 89.2%, especificidad 89.8%, valor predictivo positivo 96.1%, valor predictivo negativo 98.1%); no es útil en el diagnóstico primario de la lesión y la punción debe ser en el centro del tumor.14,19
Respecto a los auxiliares diagnósticos, la TAC y la RMN son las principales modalidades diagnósticas.6 La RMN con gadolinio es el estudio de elección para tumores de partes blandas, ya que determina su localización exacta así como las relaciones anatómicas, además de distinguir el tumor del tejido sano, teniendo como imagen característica la apariencia nodular heterogénea de bajo a ligeramente alto realce en SI en imágenes T1, heterogéneamente de bajo a alto realce en SI en imágenes T2 y reforzamiento heterogéneo posterior al contraste.20 La TC es también útil aunque es más sensible para valorar compromiso óseo.
Entre los estudios de extensión y útiles para estatificación están la radiografía de tórax para descartar metástasis pulmonares y USG de abdomen para descartar metástasis hepáticas. En caso de sospecha de metástasis se solicitan además TC de tórax, abdominopélvica o de cráneo y gammagrama óseo.14,15
Para la estatificación se sigue el sistema propuesto en 2010 por el AJCC, el cual tiene significación desde el punto de vista pronóstico al documentarse que las metástasis son regionales o a distancia. La lesión se clasifica de acuerdo al TNM, así: Tx: Tumor primario no valorable, T0: Sin evidencia de tumor primario, T1: Tumor de 5 cm o menos en su dimensión máxima; éste se subclasifica en T1a: Tumor superficial, T1b: Tumor profundo (dependiendo de si se encuentra por arriba o debajo de la fascia superficial); T2: Tumor de más 5 cm, se subdivide en T2a y T2b. N se divide en Nx: Ganglios linfáticos no valorables, N0: Sin metástasis ganglionar, N1: Metástasis ganglionar. M se divide en M0: Sin metástasis a distancia, M1: Metástasis distantes. También se toma en cuenta el grado histológico (1-3) para fines de estadificación.19,21
Respecto al tratamiento de los sarcomas de partes blandas, éste se ha revolucionado en los últimos años ya que en el pasado la cirugía radical mediante amputación era el tratamiento estándar por considerarse tumores radiorresistentes. Actualmente la tendencia es a la conservación de la extremidad gracias a la implementación de modalidades terapéuticas neoadyuvantes o posquirúrgicas. Se ha visto que la recurrencia local en pacientes que no reciben adyuvancia varía dependiendo del tipo de procedimiento quirúrgico: en caso de resección exclusivamente alcanza 90%, en resección amplia 39%, en resección de partes blandas 25% y en caso de amputación 7% a 18%.22 Por lo tanto, se ha intentado el manejo multimodal de este tipo de tumores mediante la combinación de cirugía y radioterapia, sugerida por primera vez en 1950 por Leucutia,23 habiéndose comprobado que ésta impacta el control locorregional y la supervivencia.22
En cuanto al tratamiento, en caso de tumores resecables existen dos modalidades: La resección amplia de la lesión con márgenes tridimensionales de 2 a 3 cm de tejido normal, con extirpación de la totalidad del tumor y la zona reactiva, pero sin extirpar la estructura que origina el tumor (lo cual deja cierto riesgo de tumor residual microscópico) y la resección radical en la que se extirpa el tumor completo y la estructura que lo origina (músculo, nervios, vasos); en ésta el tumor debe estar rodeado completamente de una envoltura músculofascial intacta, siendo muy baja la posibilidad de residuo microscópico.14
El tratamiento a través de radioterapia tiene varias modalidades. La radioterapia radical se otorga a pacientes cuyo tumor es irresecable por medios quirúrgicos; generalmente se administran dosis de 64-66 Gy al sitio del tumor. Este tratamiento tiene fines paliativos y el índice de control local es de 29% a 33%.14, 22
En caso de radioterapia preoperatoria se obtiene un control local de 90% a 97%. La radioterapia adyuvante se administra a pacientes sometidos a resección tumoral previa en dosis promedio de 65-79 Gy después de ocho semanas del evento quirúrgico y está indicada en casos con alto riesgo de recurrencia local, como sucede cuando hay márgenes infiltrados por tumor, pacientes sometidos a una cirugía marginal, presencia de márgenes cercanos y tumores mayores de 5 cm. La radioterapia se administra ya sea con haz de electrones o megavoltaje con fotones con fraccionamiento convencional de 1.8-2.0 Gy diariamente por cinco días a la semana con dosis total de 45-64 Gy o con hiperfraccionamiento acelerado mediante dos fracciones diarias de 1.6 Gy en intervalos de seis a ocho horas con una dosis total de 44.8 Gy o tres fracciones diarias de 1.5 Gy en intervalos de cinco horas con una dosis total de 48-54 Gy. El campo de radiación incluye el sitio del tumor inicial con 2 a 3 cm de margen de tejido sano.7 Con esta modalidad se obtiene un control local de 78% a 91%, una tasa de falla local de 18.5% y una preservación funcional del miembro de 84.7%.22 Respecto al papel de la quimioterapia en este tipo de tumores, no se ha demostrado su efectividad en el tratamiento de los mismos.7
El pronóstico de estos pacientes depende de varios factores: grado, tamaño, localización y tipo histológico del tumor. En aquellos con sarcomas grandes, profundos y de alto grado el riesgo de metástasis alcanza 40% a 50%, siendo la ruta más común la diseminación hematógena. En el caso de la variedad fibromixoide la diseminación ganglionar linfática no se presenta como acontece en otros subtipos como el sarcoma sinovial, sarcoma epiteloide y rabdomiosarcoma (afección ganglionar en 12% a 20% de los casos).14 El sitio más frecuente de enfermedad metastásica en los sarcomas de extremidades es el pulmón (35% a 50%), le siguen en frecuencia hígado (25%), hueso (22%) y encéfalo (5%). Un 30% de los pacientes sometidos a metastasectomía de lesiones pulmonares sobrevive a cinco años. Entre 20% y 30% de los pacientes tienen metástasis extrapulmonares en hígado, hueso, encéfalo.14,15 En pacientes con tumores de alto grado, el riesgo de enfermedad metastásica a distancia es de 34% en tumores de 5.1 a 10 cm, 43% en tumores de 10.1 a 15 cm y 58% en aquellos de 15.1 a 20 cm.22 La recurrencia global después de una cirugía radical oscila entre 20% y 30% y se presenta en el 80% de los casos en los dos años siguientes.14 La sobrevida global se aproxima a 17%.22 En cuanto a la sobrevida a cinco años por etapas clínicas, ésta es de 80% a 90% (etapa I), 65% (etapa II), 45% (etapa III) y 10% (etapa IV).14
La vigilancia durante los dos primeros años se realiza mediante examen físico detallado del sitio de resección cada tres meses y radiografía de tórax AP y lateral cada seis meses. Entre los tres y los cinco años se efectúa examen físico cada seis meses y radiografía AP y lateral y TAC o RMN del sitio de resección de manera anual. A partir de los cinco años un examen físico anual con radiografía AP y lateral.14
¿ CONCLUSIONES
Cabe resaltar la importancia del reporte de este caso, ya que es el primero en su tipo documentado en nuestra unidad hospitalaria; además, a nivel nacional no se han notificado casos de este subtipo de sarcomas de tejido blando. Esto crea un razonamiento a tener en cuenta en la práctica clínica y a usarlo para emitir un diagnóstico diferencial cuando se presentan tumores de este tipo. A la descripción de este caso se suma una revisión de la literatura mundial sobre las características epidemiológicas, clínicas, patológicas, diagnósticas y terapéuticas con el fin de informar detalladamente acerca de esta neoplasia de comportamiento agresivo.
¿ AGRADECIMIENTOS
Un cordial agradecimiento a todos los colaboradores que participaron en la realización de este artículo de revisión y reporte de caso: Cruz L, Cortés S, Farías MA, Tenorio JA, Ramírez JA, Conde E.
Correspondencia: Luis Cruz Benítez.
Servicio de Cirugía Oncológica, 2º piso Torre Consulta Externa; CMN "20 de Noviembre". ISSSTE.
Av. Félix Cuevas 540, Col. Del Valle, C.P. 03100, Del. Benito Juárez, México, D.F.
Teléfonos: 5200 3505, 5200 5003 Ext. 14444, 14450. Teléfono celular: 55 4135 2169.
Correo electrónico:crubeluis@yahoo.com.mx, crubeluis@gmail.com