




El síndrome de Maffucci (SM) es un padecimiento no hereditario, que se ha asociado a neoplasias no mesenquimatosas y tumores de cordones sexuales. Debido a su producción de estradiol, hasta 80% de los casos presentan pubertad precoz. Alrededor del 20% de estos pacientes desarrollan tumores, principalmente malignos, entre los que se encuentran astrocitomas, adenomas pituitarios, tumor de células de la granulosa y adenocarcinoma pancreático. Los encondromas y hemangiomas pueden presentar transformación maligna a condrosarcomas y hemangiosarcomas. Los tumores ováricos de células de la granulosa son raros, la forma juvenil se diagnostica en menores de 30 años, se presentan con masas abdominales y pubertad precoz. El pronóstico es favorable y el tratamiento de elección es la cirugía.
Se presenta femenino de 13 años de edad, con antecedente de SM. Durante el seguimiento médico presentó datos clínicos de pubertad precoz; en el perfil hormonal se encontró elevación de estradiol. La tomografía abdominal demostró un tumor dependiente de ovario. La neoplasia fue resecada completamente y el estudio histológico estableció el diagnóstico de tumor de células de la granulosa, que se clasificó como estadio IA.
El SM es una condición rara, igualmente lo son los tumores de células de la granulosa. Este caso ejemplifica la importancia de realizar un examen clínico completo en los pacientes con SM, para identificar no la trasformación maligna en los encondromas y hemangiomas, sino también tumores de ovario, pues su diagnóstico precoz es fundamental para la curación.
Maffucci syndrome (MS) is a non-hereditary condition that has been associated with mesenchymal neoplasms and sex cord-stromal tumors. Because of estradiol production, up to 80% of cases have precocious puberty. About 20% of these patients develop tumors, particularly malignant, among which are: astrocytomas, pituitary adenomas, granulosa cell tumors and pancreatic adenocarcinoma. Enchondromas and hemangiomas may present malignant transformation to chondrosarcoma and hemangiosarcomas. Ovarian granulosa cell tumors are rare, the juvenile form is diagnosed in patients less than 30 years of age, they present with abdominal masses and precocious puberty. The prognosis is favorable and surgery is the treatment of choice.
A 13 year-old girl, with a history of MS. During medical follow-up precocious puberty was detected and the hormonal profile detected estradiol elevation. Abdominal tomography showed an ovarian tumor. The tumor was completely resected and histology established the diagnosis of granulosa cell tumor, which was classified as stage IA. Mafucci syndrome is a rare condition and so are granulosa cell tumors. This case illustrates the importance of performing a comprehensive clinical examination in MS patients to identify not only the malignant transformation of enchondromas and hemangiomas, but also other tumors, since early diagnosis is essential for the outcome.
Introducción
El síndrome de Maffucci (SM) se describió en 1881, por Angelo Maffucci. Es una enfermedad no hereditaria que se caracteriza por la presencia de encondromas múltiples de los huesos largos, hemangiomas de tejidos blandos y en ocasiones linfangiomas. Es uno de los subtipos más frecuentes de las encondromatosis, que son trastornos raros, heterogéneos, presentes en el esqueleto, donde se encuentran múltiples encondromas1,2.
Los encondromas son neoplasias benignas que forman cartílago hialino en la médula de los huesos, son generalmente asintomáticas y surgen frecuentemente en las metáfisis y diáfisis de los huesos tubulares cortos y largos de las extremidades, especialmente en manos y pies. Por lo general, ocurren como una lesión simple que se detecta incidentalmente por estudios radiográficos. En ocasiones los pacientes presentan múltiples encondromas, las lesiones muestran distribución asimétrica y pueden causar deformidad severa de los huesos. En el 25% de los casos, la enfermedad se desarrolla desde el nacimiento o en el primer año de vida, en el 45% inicia antes de los 6 años y en el 78% de los pacientes se diagnostica antes de la pubertad. Muestra ligero predominio por el género femenino3.
El SM se ha asociado a tumores no mesodérmicos, entre los que se encuentran los astrocitomas, los adenomas pituitarios, el tumor de células de la granulosa y el adenocarcinoma pancreático. Los encondromas y hemangiomas pueden presentar degeneración maligna a condrosarcomas y hemangiosarcomas. Los hemangiomas son tumores benignos vasculares, que frecuentemente sobresalen como nódulos blandos azules o rojizos, y pueden encontrarse en cualquier parte del cuerpo.
Los tumores ováricos de las células de la granulosa son neoplasias raras, que constituyen sólo 2% a 5% de los tumores ováricos. Tienen una incidencia de 0.58 a 1.6 casos/100,000 mujeres por año. Existen 2 subgrupos de acuerdo a su presentación clínica y características histológicas: la forma adulta y la juvenil. Esta última comprende solamente el 5% del total de casos y se diagnostica en las primeras 3 décadas de la vida, con mediana de 8 a 9 años cuando se incluyen sólo menores de 16 años, y de 13 a 17 años cuando las series incluyen todas las edades4. La mayoría de las pacientes se presenta con dolor abdominal localizado, distensión abdominal y masa palpable en pelvis o abdomen. El 10% de los casos puede debutar con abdomen agudo, debido a ruptura del tumor y hemoperitoneo o por torsión del mismo. La mayoría de las pacientes prepúberes tienen pubertad precoz isosexual, caracterizada por desarrollo prematuro de genitales externos, crecimiento de vello púbico y axilar, secreción vaginal y hemorragia uterina. El tumor es bilateral en 3% de los casos, generalmente está limitado al ovario al momento del diagnóstico5,6.
Reportamos un caso de tumor de células de la granulosa juvenil del ovario, en una niña con SM.
Presentación del caso
Femenino de 13 años de edad, originaria y residente del estado de Hidalgo. Producto de la tercera gestación, sin control prenatal; nació a término por parto eutócico, llanto y respiración espontáneos, con peso bajo al nacer (1,600 g). Retraso en el desarrollo psicomotor, manifestado por sostén cefálico al 9º mes, bisílabos a los 18 meses, marcha a los 3 años y control de esfínteres a los 3-4 años.
En el año 2003, presenta aumento de volumen y lesiones en tejidos blandos en extremidades superiores e inferiores y región torácica, de consistencia blanda, color rojo y deformidades óseas. En el examen físico se encontró con estatura por debajo del percentil 50 para la edad y género, deformidad de extremidades superiores y lesiones vasculares de partes blandas (fig. 1). Se integró el diagnóstico de SM.

Figura 1. Fotografía clínica que muestra el fenotipo de la paciente, con talla por debajo del percentil 50, deformidades de las extremidades superiores y hemangiomas.
En julio de 2004, se realizó resección de malformaciones vasculares de ambas manos (fig. 2). El reporte histopatológico fue de hemangiomas cavernosos.

Figura 2. Hemangiomas y deformidad de las extremidades superiores.
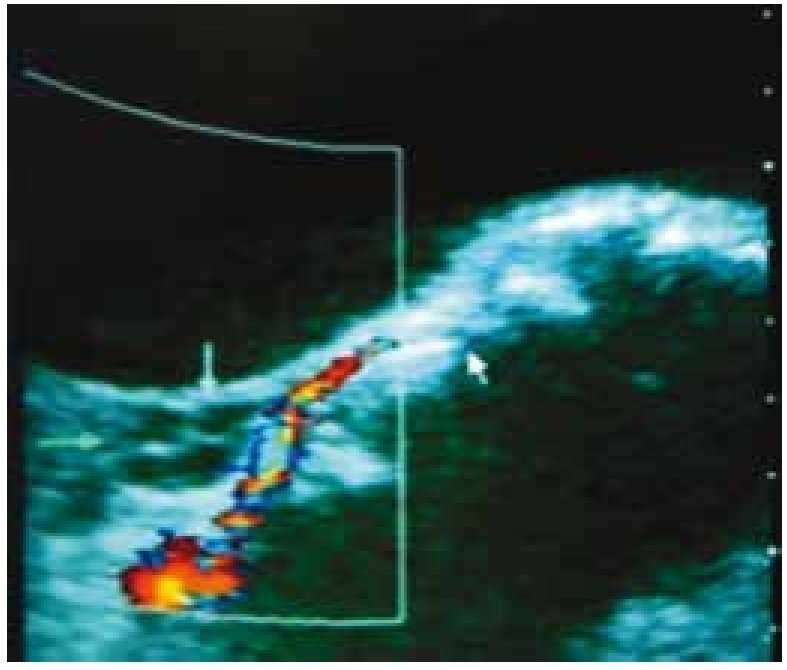
Inicia menarca en noviembre de 2009, con ciclos irregulares y polimenorrea, Tanner mamario II, Tanner de vello púbico II. En diciembre de 2009, se detecta durante la exploración física una masa abdominopélvica. El perfil hormonal reportó: estradiol 671 pg/mL, hormona luteinizante (LH) < 0.10 mU/mL, hormona folículo estimulante (FSH) < 0.10 mU/mL, hormona adrenocorticotropa (ACTH) 33.7 pg/ mL, cortisol 11 mg/dL, prolactina 22.4 ng/mL, alfa-fetoproteína sérica (AFP) 4.6 UI/mL y fracción beta de hormona gonadotropina coriónica humana (FBHGC) 3.8 UI/m. Se sospecha de un tumor ovárico, se realiza ultrasonido (USG) donde se encuentra masa pélvica heterogénea de 135 × 62 × 133 mm, con vascularización importante (fig. 3).
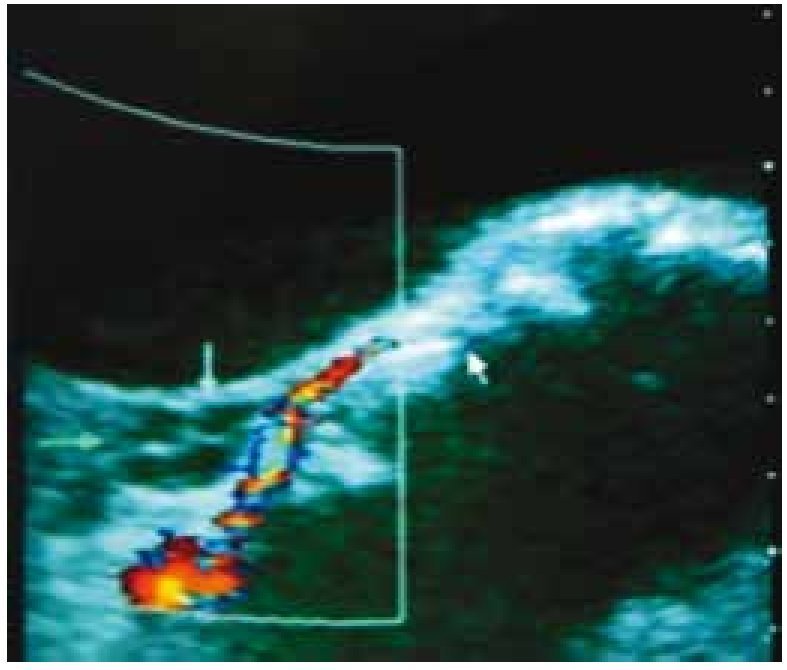
Figura 3. Ultrasonografía realizada en diciembre de 2009, en la cual se observa masa pélvica heterogénea de 135 × 62 × 133 mm, con vascularización importante.
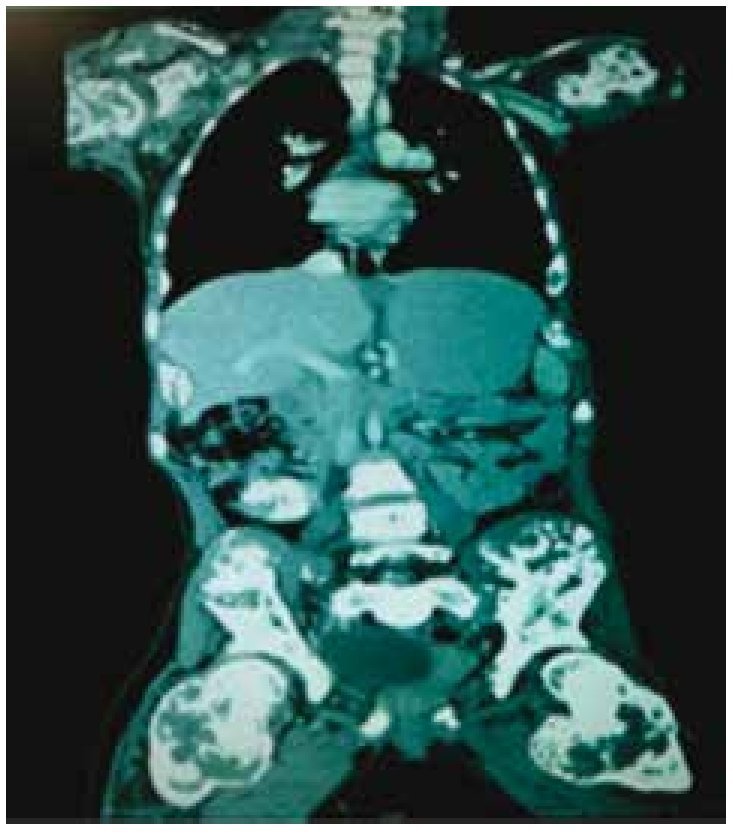
La tomografía computada mostró una lesión en fosa ilíaca derecha y flanco derecho que se extendía hacia la línea media, predominantemente quístico, con medidas de 16.3 × 14.2 cm en diámetros transverso y longitudinal, que desplazaba útero, vejiga urinaria y vena cava inferior, el útero con líquido en endometrio, además de evidenciarse encondromatosis en múltiples huesos (fig. 4).
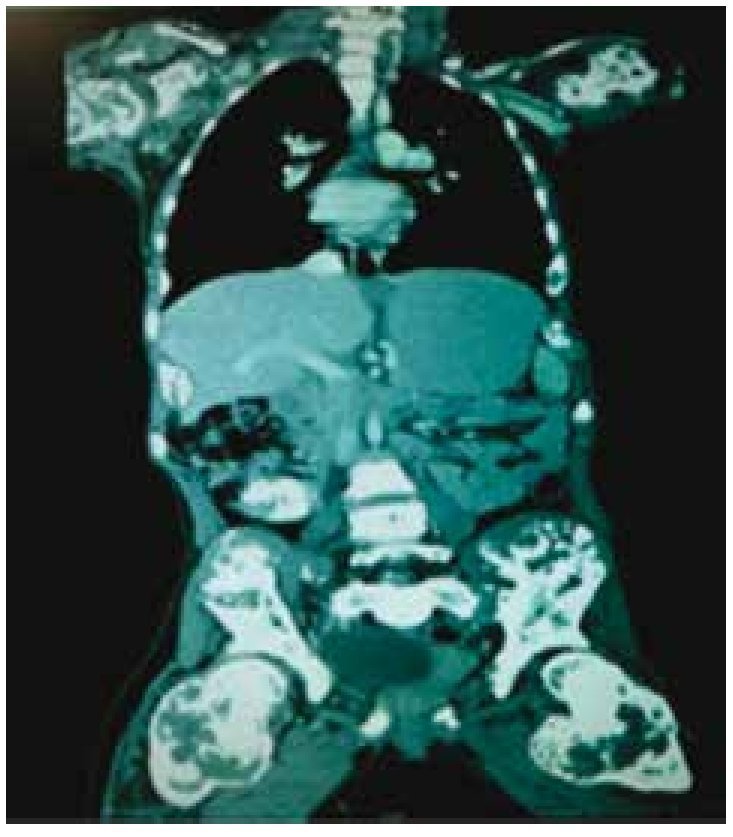
Figura 4. Tomografía computada en corte coronal, donde se identifican tumor intrapélvico y encondromas en múltiples huesos.
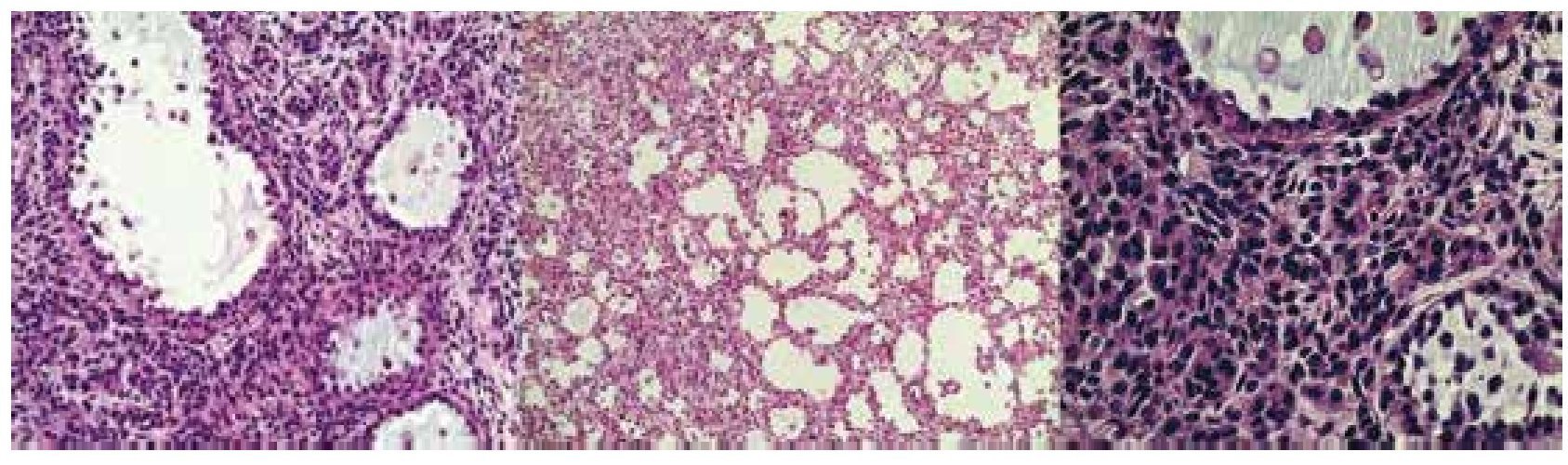
El 11 de diciembre de 2009 se somete a laparotomía, encontrándose tumor dependiente de ovario izquierdo y 2 quistes paratubáricos derechos. El reporte de patología fue: Tumor de células de la granulosa quístico (fig. 5), salpinge izquierda sin tumor, en región paratubaria hidátide de Morgagni, correderas cólicas, diafragma derecho e izquierdo y epiplón sin neoplasia. La paciente se encuentra actualmente viva, libre de enfermedad, a poco más de 3 años de seguimiento.
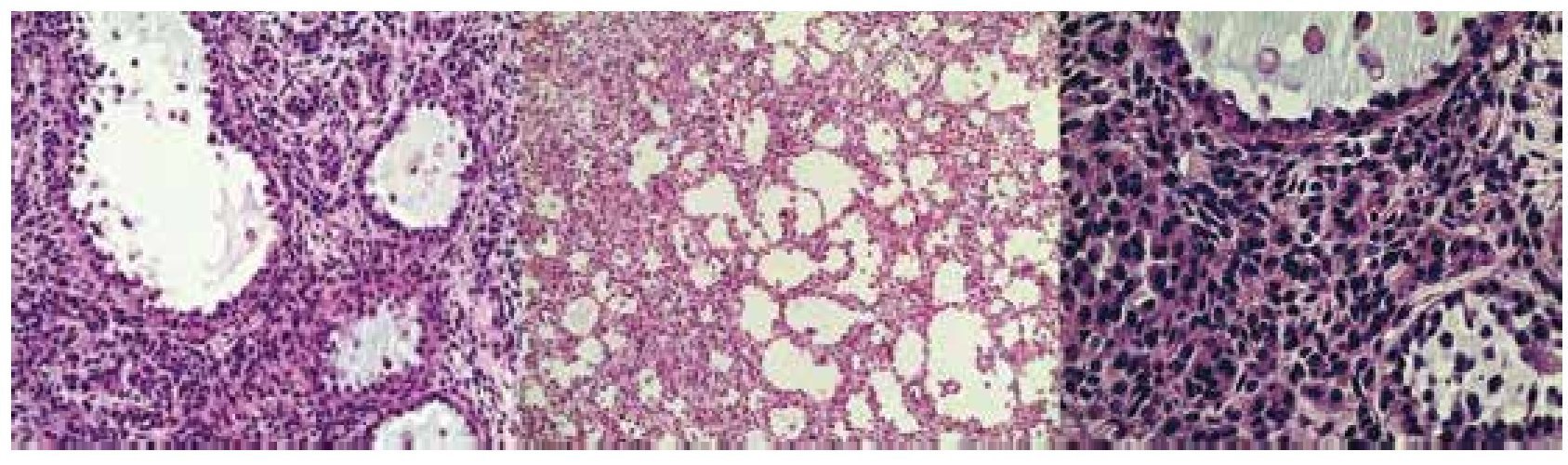
Figura 5. Imagen histológica del tumor ovárico. Los cortes muestran células de mediano tamaño con escaso citoplasma eosinófilo, núcleo ovoide y nucléolos prominentes, con hendiduras (granos de café).
Discusión
La asociación entre SM y tumores ováricos ha sido descrita con anterioridad, existen aproximadamente 200 casos reportados de SM en la literatura médica; su asociación con tumor del estroma ovárico es de tan sólo 10 casos, aproximadamente. De un total de 5 casos de tumores de la granulosa tratados en el Hospital Infantil de México Federico Gómez en un lapso de 10 años, éste es el único caso que muestra esta asociación.
Los tumores no epiteliales malignos del ovario son cánceres raros, cuya historia natural no es bien conocida y para los cuales los factores pronósticos continúan estudiándose. Los tumores del estroma y de los cordones sexuales se originan del tejido conjuntivo del ovario, representan aproximadamente 5% de las neoplasias de ovario en el paciente pediátrico, debido a que estas células participan en la función ovárica hormonal. La mayoría de los tumores del estroma o de los cordones sexuales son capaces de secretar hormonas (estrógenos, andrógenos y corticoides), lo cual explica la pubertad precoz asociada con estos tipos de cáncer. Estos tumores típicamente se detectan en etapas tempranas y pueden recurrir hasta 30 años después del tratamiento inicial. Pueden presentarse como única histología o en combinación con otros tipos histológicos.
Los tumores de las células de la granulosa juvenil son tumores del estroma gonadal que se presentan más frecuentemente en niñas, adolescentes y adultas jóvenes, representando aproximadamente el 10% de todas las neoplasias del ovario en niñas. Difieren de la forma adulta no sólo por la edad de presentación, sino por las características histológicas y clínicas. Histológicamente, se distinguen de la forma adulta por los núcleos generalmente redondeados e hipercromáticos, citoplasma eosinófilo y presencia de vacuolas.
Cuando la variante juvenil se presenta antes de la pubertad, se acompaña de manifestaciones de pubertad precoz, además de los signos y síntomas asociados al crecimiento del tumor. Se caracteriza por crecimiento lento e indolente, dando lugar a tumores de gran tamaño al momento del diagnóstico.
Se ha encontrado asociación con algunos síndromes genéticos, entre ellos se encuentran el síndrome de Potter, enfermedad de Ollier y el SM.
La diseminación del tumor es generalmente local, por extensión directa y siembras intraperitoneales, sin embargo puede también diseminarse por vía hematógena y dar lugar a metástasis a pulmón, hígado y cerebro, aunque cuando esto ocurre, es generalmente años después del diagnóstico.
Tanto la forma juvenil como la adulta son neoplasias menos agresivas que los cánceres ováricos epiteliales, tienen la tendencia a diagnosticarse en estadios localizados y por lo general, son de buen pronóstico.
El estadio es el factor pronóstico que se relaciona en forma inequívoca con recurrencia. El tumor de las células de la granulosa tiene una supervivencia global a 5 años de 90% y a 10 años de 60% a 90%. La tasa de supervivencia global a 3 años para etapas IA y IB en la variante juvenil, se estima en 97%. Otros factores pronósticos que se mencionan como significantivos son la ruptura del tumor, aunque según las series los resultados son variables7-9. Los factores histológicos que se relacionan con el pronóstico son el índice mitótico (< 5 mitosis por 10 campos de alto poder), con supervivencia global a 10 años de 70% vs. 37% en los que tiene mayor número de mitosis10, y la atipia nuclear se considera un factor predictor de recurrencia tumoral11. El tratamiento de estos tumores es quirúrgico y dado el excelente pronóstico y las etapas tempranas en las que comúnmente se detecta, es poco frecuente que se emplee quimioterapia o radioterapia, reservándose dichas opciones para casos recurrentes o para aquellos que tienen características de mal pronóstico (atipia nuclear, ruptura del tumor, índice mitótico > 5 mitosis en 10 campos de alto poder y aneuploidía).
El riesgo de que existan tumores malignos en los pacientes con SM, puede llegar a ser de hasta 25%, siendo la mayoría de ellos tumores mesenquimatosos. La asociación entre encondromatosis y tumores de células de la granulosa ha sido documentada previamente, aunque no se conoce el gen que pudiese ser el causal12.
Es importante destacar el hecho de que en los pacientes prepúberes con datos de pubertad precoz, se debe considerar la posibilidad de una causa neoplásica, ya que esta es una forma de presentación de tumores que secretan hormonas sexuales o gonadotropinas, tanto gonadales como extragonadales, como en el caso de los tumores de células de la granulosa variedad juvenil. Como estudio de diagnóstico precoz, se propone el ultrasonido como primera opción, que es suficientemente sensible para la identificación de tumores intrapélvicos gonadales.
Financiamiento
No se recibió ningún patrocinio para llevar a cabo este artículo.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
* Autor para correspondencia:
Dr. Márquez N° 162, Colonia Doctores, Delegación Cuauhtémoc, C.P. 06720, México D.F., México.
Teléfono (oficina): 5522 8899, ext. 2124. Celular: (044 55) 3460 4505.
Correo electrónico: phalomi@hotmail.com (Miguel Ángel Palomo-Colli).