


PRESENTACIÓNDELCASO
Se presenta el caso de un joven de 17 años que tenía cuadro clínico de un año de evolución, caracterizado por astenia, aumento de volumen a nivel plantar izquierdo, sin dolor inicial y luego a nivel de cuello con adenopatías bilaterales, pérdida de peso no cuantificada, náuseas y dolor abdominal. Se practicó biopsia de adenopatía cervical, en la que se detectó metástasis de carcinoma poco diferenciado. Después de un segundo estudio de biopsia, el informe histopatológico indicó tumor desmoplásico de células pequeñas y redondas, con los siguientes datos de inmunohistoquímica: CD25 negativo, CK negativo, CK18 negativo, AME inmunorreactividad positiva, CD117 negativo, Masson negativo, PAS positivo.
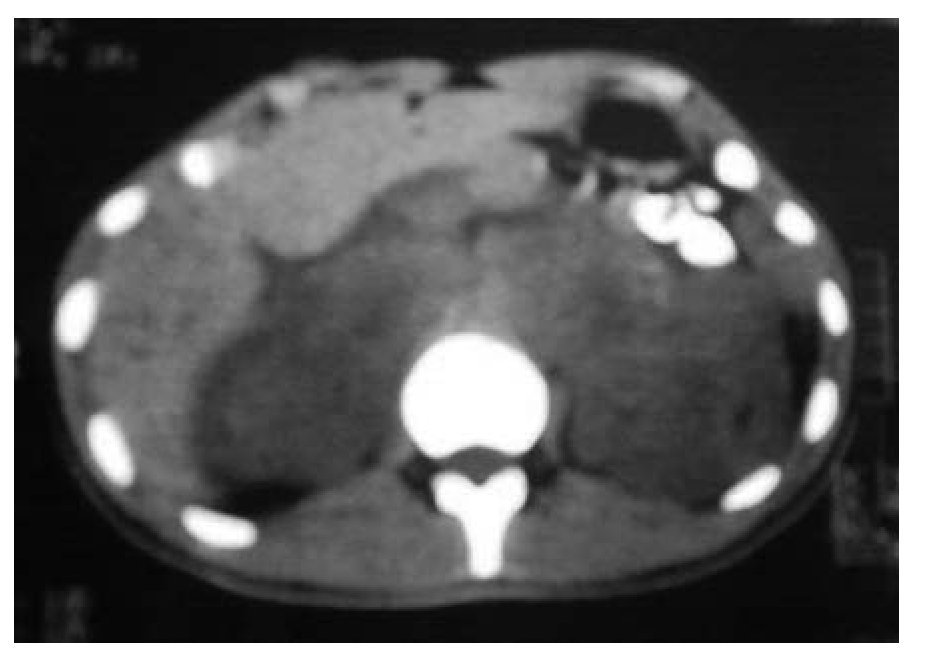
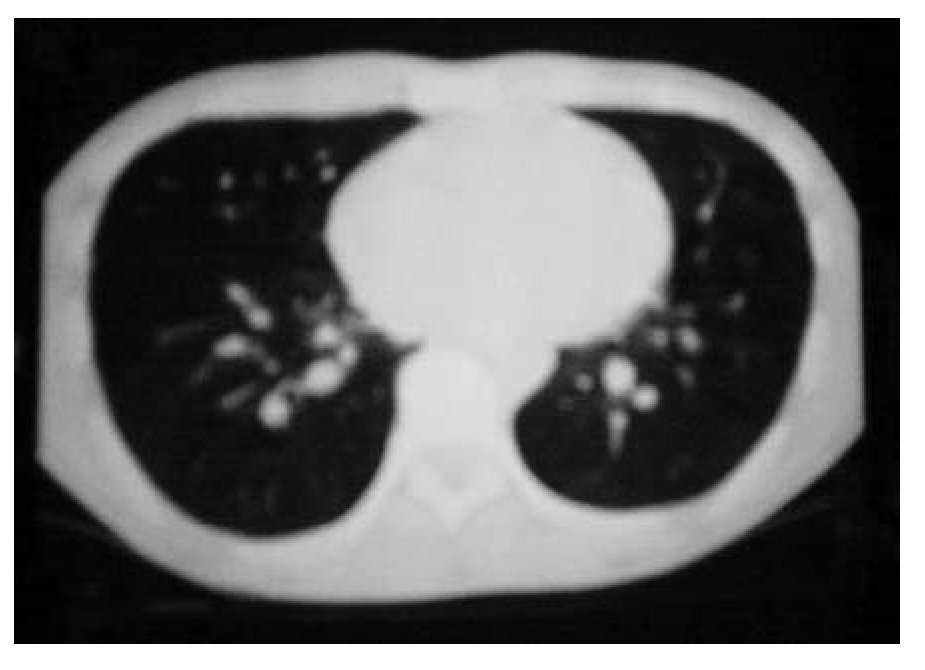
Se tomó gamagrama con galio, en el que hubo hipercaptación en los niveles supraclavicular izquierdo y mediastínico, lo mismo que en colon descendente, tibia izquierda y talón derecho. Se confirmó el diagnóstico por biopsia de tejidos blandos del pie. Se realizaron tomografía de tórax con imagen paracardiaca derecha (Figura 1A) y tomografía de abdomen, con lo que se detectaron nódulos perivertebrales en ambos lados, en polo superior de riñón (Figura 2A).
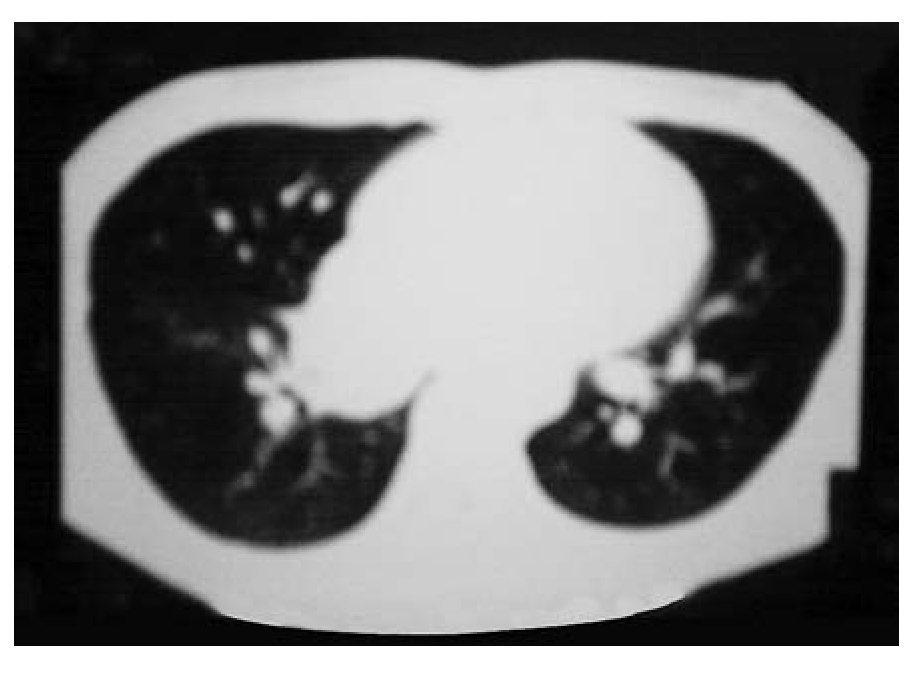
Figura 1A. Imagen paracardiaca derecha.
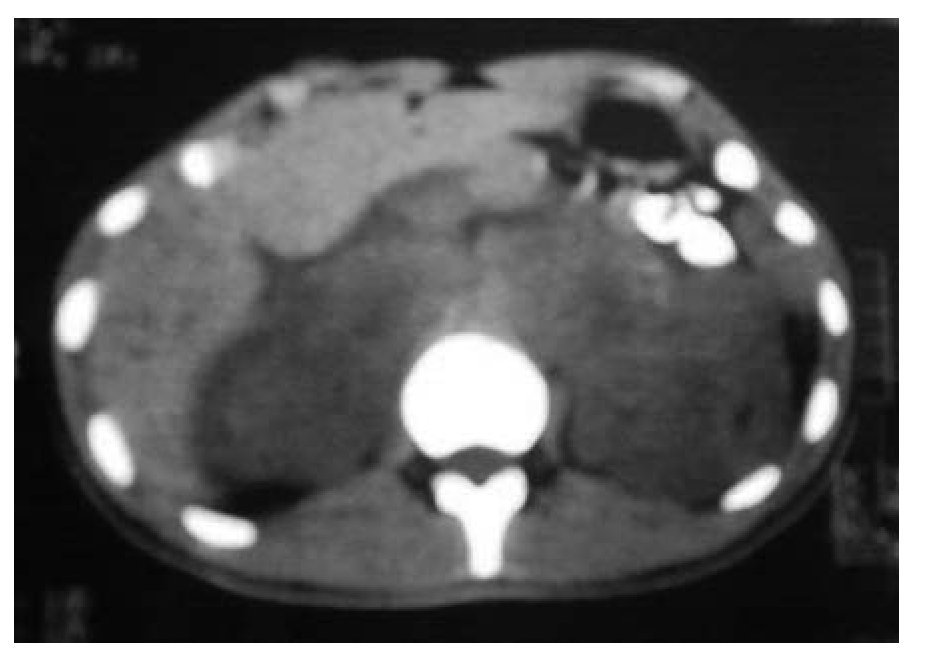
Figura 2A. Nódulos perivertebrales en ambos lados del polo superior de riñón.
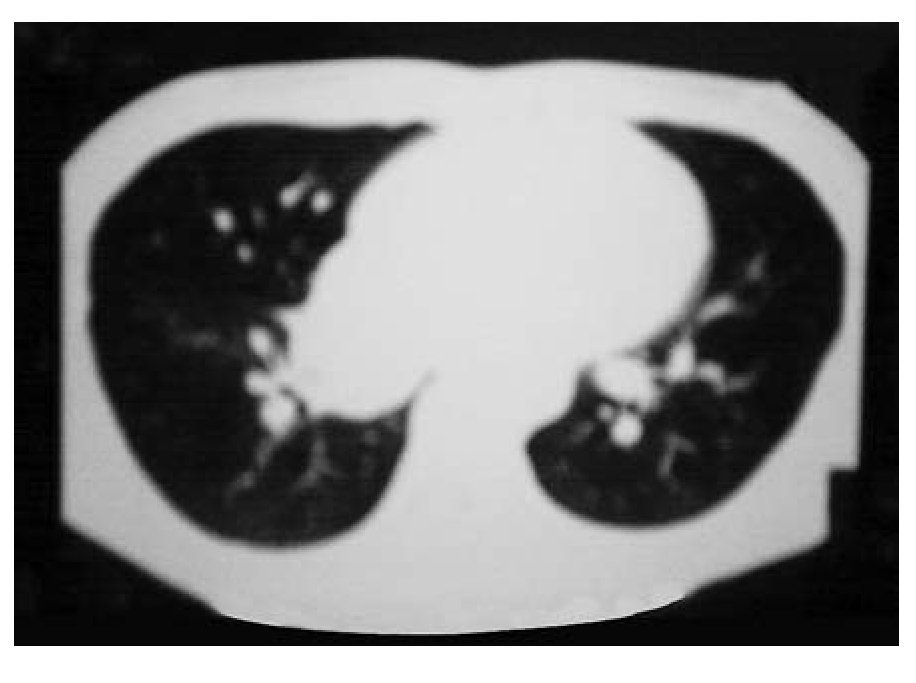
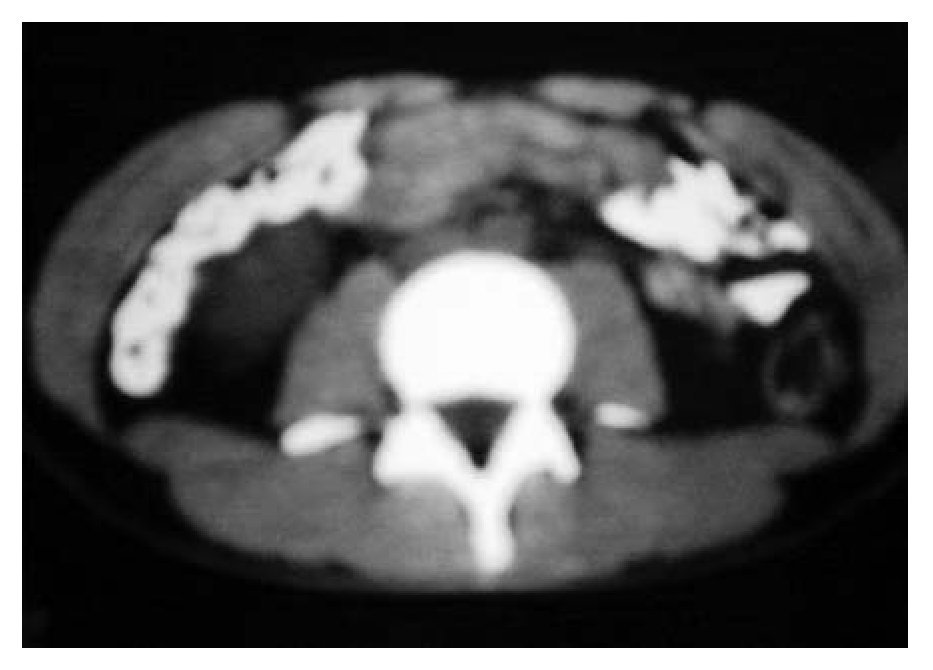
Se inició un esquema de quimioterapia basado en administración de ciclofosfamida (500 mg/m2 SC), adriamicina (25 mg/m2 SC), etopósido 80 mg/m2 SC, días uno a tres) y bevacizumab (2.5 mg/kg/semana) cada 21 días, durante seis ciclos. Al finalizar el tratamiento, se llevaron a cabo estudios de tomografía, pero no se observaron signos de actividad tumoral (Figura 1B y 2B). Un mes después, se desarrolló un cuadro de insuficiencia cardiaca y, con base en un estudio de ecocardiografía, se diagnóstico miocardiopatía dilatada. Se inició tratamiento con digoxina, metoprolol y furosemida. Se realizó rastreo óseo que dio resultado negativo, con informe de hipercaptación a nivel dorsal del pie izquierdo. Por medio de tomografía de pie, se detectó un tumor de 6 cm a este nivel. Se inició radioterapia paliativa en el pie, con dosis de 30 Gys. Más tarde se desarrolló adenopatía inguinal izquierda; se tomó muestra para biopsia y el estudio dio resultado positivo de neoplasia maligna de células pequeñas redondas y azules, poco diferenciada. Luego se obtuvo resultado positivo en un rastreo óseo con galio, a nivel de hombro, clavícula derecha, mediastino, lóbulo derecho de hígado, mesogastrio, ganglio inguinal izquierdo, fémur, tibia y tobillo izquierdo. El estado funcional del paciente se deterioró, por lo que sólo se le proporcionó tratamiento de sostén. Ocho meses después de haber concluido la quimioterapia, falleció por insuficiencia cardiaca descompensada.
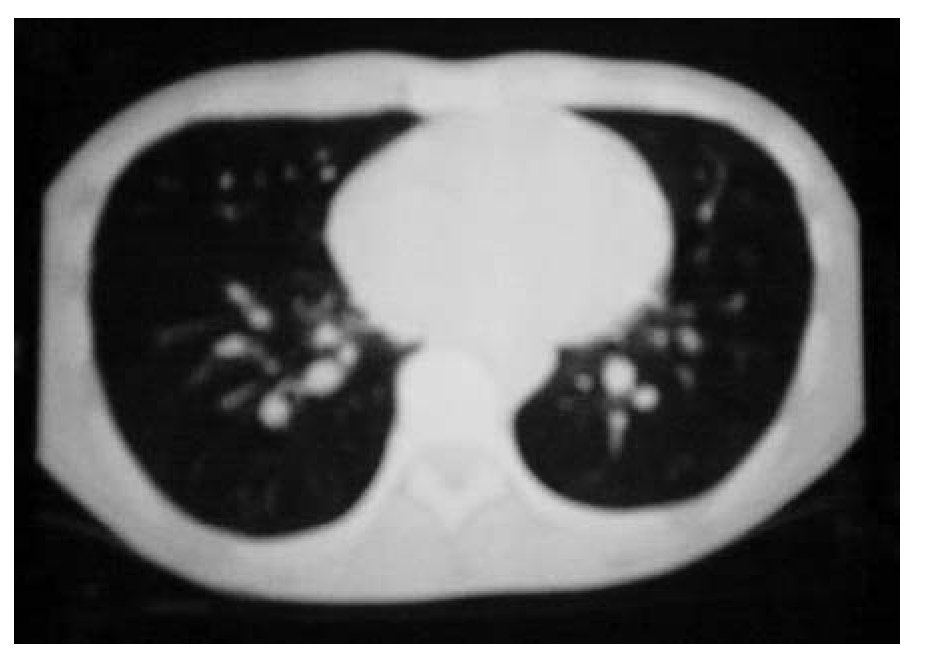
Figura 1B. Respuesta completa al tratamiento.
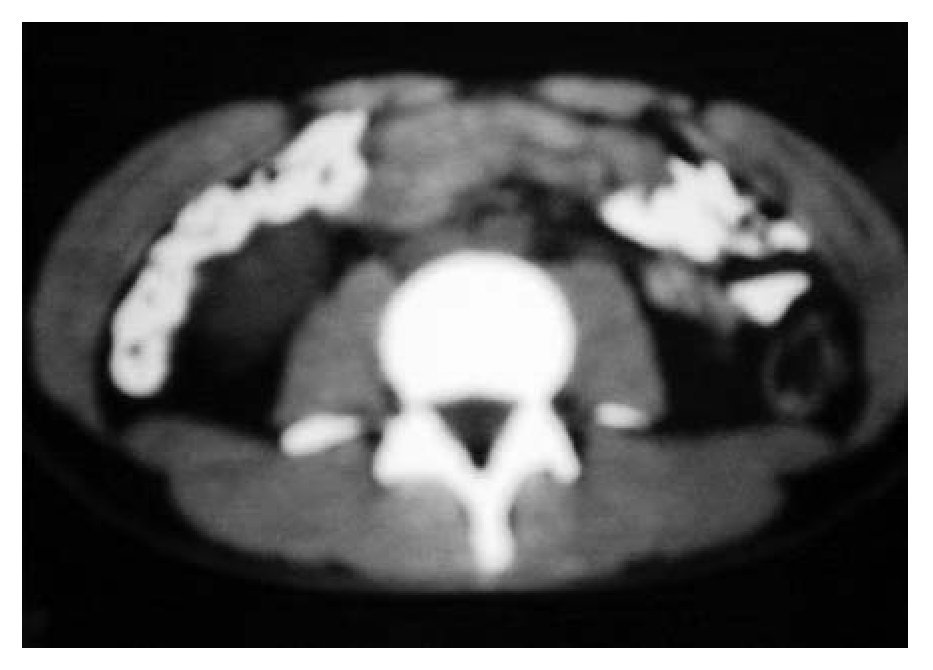
Figura 2B. Respuesta completa al tratamiento.
DISCUSIÓN
El tumor desmoplásico de células redondas pertenece a la familia de "tumores de células pequeñas redondas y azules", formada por neuroblastoma, linfoma, rabdomiosarcoma, sarcoma de Ewing, tumor de Wilms y tumor neuroectodérmico primitivo (PNET). Afecta sobre todo a los adolescentes y adultos jóvenes. Predomina en varones, en una relación 3.8:1. La edad media al momento del diagnóstico es 14.5 años, aunque se ha descubierto en personas de edad más avanzada. Con frecuencia se manifiesta con un cuadro de distensión y malestar abdominal inespecífico, estreñimiento y pérdida de peso, a nivel de cavidad abdominal, pelvis y órganos viscerales, y causa procesos obstructivos.1 Los signos tomográficos más comunes a nivel abdominal son masas múltiples de tejidos blandos, en epiplón, mesenterio o superficie peritoneal, sin que se distinga su origen en algún órgano; además se aprecian calcificaciones de la tumoración, metástasis hepáticas, adenopatías, ascitis, obstrucción urinaria o intestinal.2
Gerald y Rosai describieron por primera vez (en 1989) el tumor desmoplásico de células redondas, con características histológicas específicas de inmunohistoquímica y cariotipo.3 Antes, se clasificaba a este tipo de tumores como una variedad atípica de tumores de células redondas y pequeñas. Una de las características histológicas de dichas neoplasias es el estroma desmoplásico que envuelve las células tumorales. Por lo general, este estroma es colágeno condensado o fibromixoide que reviste nidos bien definidos de células primitivas no diferenciadas. El tumor es reactivo a marcadores epiteliales, mesenquimatosos y neurales, lo que es una característica útil para distinguirlos de PNET, linfomas, rabdomiosarcomas y otros tumores pediátricos con aparición de células redondas pequeñas y azules.
El diagnóstico histopatológico inicial puede incluir la realización de una BAAF, aunque, debido a las características de las células que conforman este grupo de tumores, se requiere de una adecuada cantidad de tejido para realizar las pruebas de inmunohistoquímica; cuando existe la neoplasia, los exámenes dan resultados positivos de citoqueratina, antígeno epitelial de membrana y desmina, aunque a veces también se detecta reactividad a vimentina, proteína S100 y enolasa específica de neurona. Se considera que son tumores de moderada y alta celularidad.4,5
En cuanto al cariotipo de estas neoplasias, se observa una translocación característica en los cromosomas 11 y 22, t(11;22)(p3;q12), que abarca dos regiones cromosómicas que también participan en el desarrollo de otros tumores malignos. La banda 22q12 es el sitio de EWS, un gen alterado con translocaciones cromosómicas específicas de tumor y 11p13 es el sitio genético del tumor de Wilms, WT1. En varios estudios se ha establecido que estos dos genes están funcionalmente fusionados en los tumores desmoplásicos de células redondas y son fundamentales para la oncogenia.6,7
Aún se desconoce su histopatogenia. Como se forman en la cavidad peritoneal y carecen de actividad tumoral visceral primaria de origen, se cree que el tumor nace en mesotelio o en tejido submesotelial o subseroso mesenquimatoso. A casi 20 años de la descripción inicial de este grupo de tumores, su diagnóstico y tratamiento aún se dificultan a patólogos y clínicos, aunque con los estudios de inmunohistoquímica se ha logrado un avance en el diagnóstico correcto.8
Se ha informado que en algunos casos se administró quimioterapia de dosis altas a pacientes con diagnóstico inicial de rabdomiosarcoma; se utilizó un esquema terapéutico con vincristina, ifosfamida y actinomicina D (IVA) y vincristina, carboplatino y tenipósido (VINCAEPI), sin lograr una respuesta adecuada; también se administró etopósido y cisplatino o 5 fluorouracilo y estreptozocina como tratamiento de segunda línea, sin obtener respuesta; luego se practicó resección quirúrgica de la neoplasia, sobre todo si estaba en posición abdominal o retroperitoneal.
Otro esquema utilizado es con ciclofosfamida, etopósido, doxorrubicina, cisplatino (esquema PAVEP) o con epirrubicina (PEVEP); hay informes de cuatro casos en que este esquema terapéutico, aplicado durante cuatro a nueve meses, permitió estabilizar la enfermedad. También se ha utilizando tratamiento de rescate con paclitaxel, dacarbazina e interferón sin obtener beneficios e, incluso, se aplicó radioterapia más 5 fluorouracilo como radiosensibilizador, sin lograr una respuesta adecuada.9 También se trató a un grupo de 12 pacientes con un esquema de dosis altas, protocolo P6, que incluye siete ciclos de quimioterapia alterna con altas dosis de ciclofosfamida, doxorrubicina y vincristina, además de ifosfamida y etopósido, así como tratamiento posquimioterapia con radioterapia, régimen mieloablativo con tiotepa, carboplatino y rescate de células madre; con este esquema, en plazo de nueve a 33 meses se logró la remisión completa en siete pacientes que, sufrían enfermedad progresiva.10
Se ha informado que los aumentos en la dosis de quimioterapia ha ocasionado neutropenia (grado 4) hasta en 91% de los casos, neutropenia febril en 68% y trombocitopenia en 68%; después se practicó resección quirúrgica o radioterapia (o ambas).11 Con quimioterapia de altas dosis se han logrado respuestas globales hasta en 43% de los casos; sin embargo, a pesar del resultado obtenido con la quimioterapia de altas dosis, estos tumores son quimiorresistentes.12
Bevacizumab es un anticuerpo monoclonal humanizado contra el factor de crecimiento de endotelio vascular (VEGF) y se ha demostrado que en combinación con quimioterapia es activo contra varios tipos de neoplasias. El uso de bevacizumab más doxorrubicina en estudios de fase 2, en pacientes con diagnóstico de sarcoma, ha generado una tasa de respuesta de 12%, no mayor que la observada con doxorrubicina como agente único y con mayor cardiotoxicidad de grado 2 hasta en 35% de los pacientes.13 Además, se detecta mayor incidencia de trastornos tromboembólicos, como infarto de miocardio y accidente vascular cerebral, por lo que es necesario utilizar estos fármacos con precaución en pacientes que tienen antecedentes de angina y cardiopatía isquémica.14
En un estudio de fase II con pacientes que tenían diagnóstico de cáncer mamario se utilizó bevacizumab como monoterapia; dos de 75 pacientes sufrieron insuficiencia cardiaca congestiva, aunque ambos habían recibido tratamiento previo con antraciclinas y radioterapia a pared torácica. No se ha dilucidado cómo se relaciona la disfunción cardiaca con la exposición a antracicinas y el tratamiento con bevacizumab.15
En estudios de pacientes con diagnóstico de cáncer de mama, se ha informado riesgo de cardiotoxicidad por el tratamiento con bevacizumab, con incidencia de cardiopatía congestiva de grado 3 de 2.2%, en comparación con un riesgo de 0.5% en pacientes que sólo reciben quimioterapia; el bevacizumab también conlleva riesgo de 0.4% de miocardiopatía de grado 3 y de 0.4% de miocardiopatía de grado 4, mientras que quienes reciben sólo quimioterapia tienen 0.5% de riesgo de miocardiopatía de grado 4.16
CONCLUSIONES
No se cuenta con un tratamiento estándar para tumores desmoplásicos de células redondas y pequeñas, de modo que se utilizan diversas combinaciones de quimioterapia. El esquema descrito en este informe de caso genera respuestas adecuadas pero fugaces; además, se debe utilizar con precaución la combinación de bevacizumab con antraciclinas, debido a los informes de incremento en la cardiotoxicidad.
Correspondencia:
Dra. Carolina Durán G.
Boulevard Morelos 340 Col. Bachoco 2º Piso Módulo E. C.P. 83143. Teléfono 01 (662) 109 05 00 / 109 05 23.
Correo electrónico: draduran@hotmail.com