




El síndrome de Bean o síndrome de nevus azul en tetina de goma es una enfermedad congénita rara consistente en malformaciones venosas multifocales localizadas predominantemente en piel, tejidos blandos y tracto gastrointestinal.
Presentamos el caso de una paciente de 15 años de edad, natural de Marruecos, que aporta informes con antecedentes de «hemangiomatosis difusa» y necesidad transfusional mensual desde los 6 meses de edad. Como antecedentes quirúrgicos refiere intervención por invaginación abdominal en julio de 2011 y una neurocirugía a los 4 años de edad sin saber precisar el motivo de la misma. Antecedentes familiares sin interés.
Acude a nuestro hospital con cuadro de 24 h de evolución de intensa astenia, cefalea y malestar general, sin dolor ni datos de sangrado externo, en relación con anemia grave sintomática (hemoglobina 5,1g/dl, hematocrito 16,5%, VCM 70 fl). Ingresa en el servicio de pediatría, tras transfusión inicial en urgencias de 2 concentrados de hematíes. En la exploración física la paciente se encuentra hemodinámicamente estable, afebril, con palidez mucocutánea y presencia de varias lesiones angiomatosas en superficie corporal de unos 5-7mm (en planta de pie derecho, antebrazo izquierdo, muslo izquierdo y región glútea derecha). Soplo sistólico panfocal. Resto normal por aparatos.
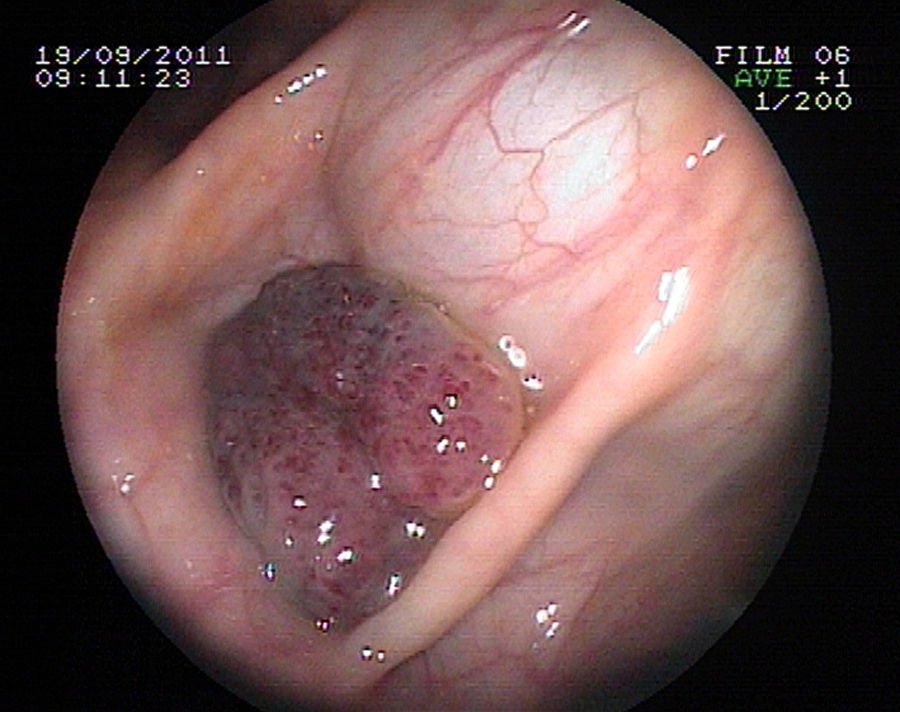
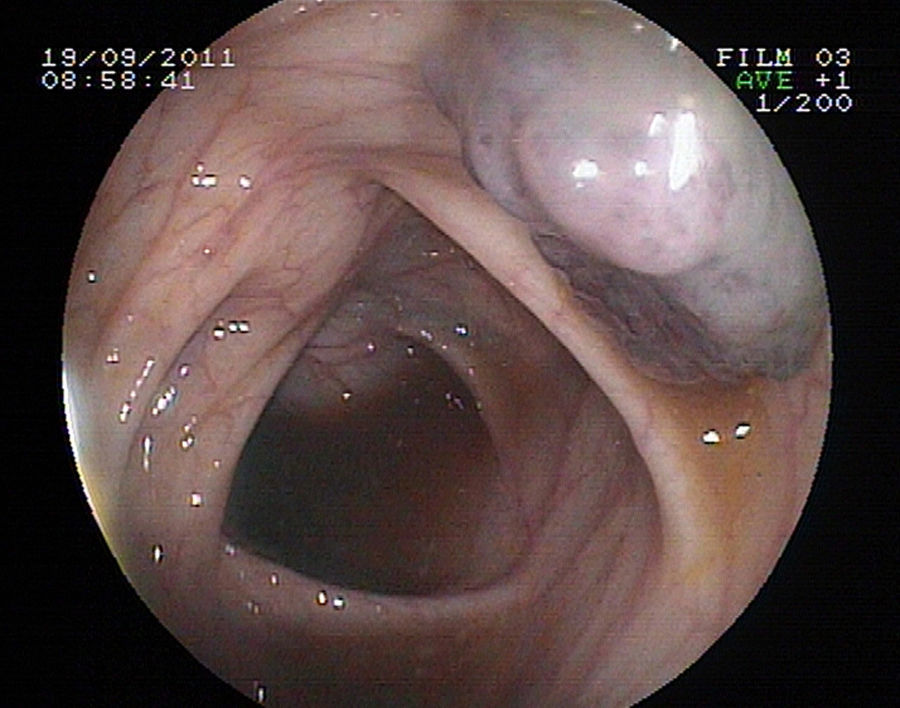
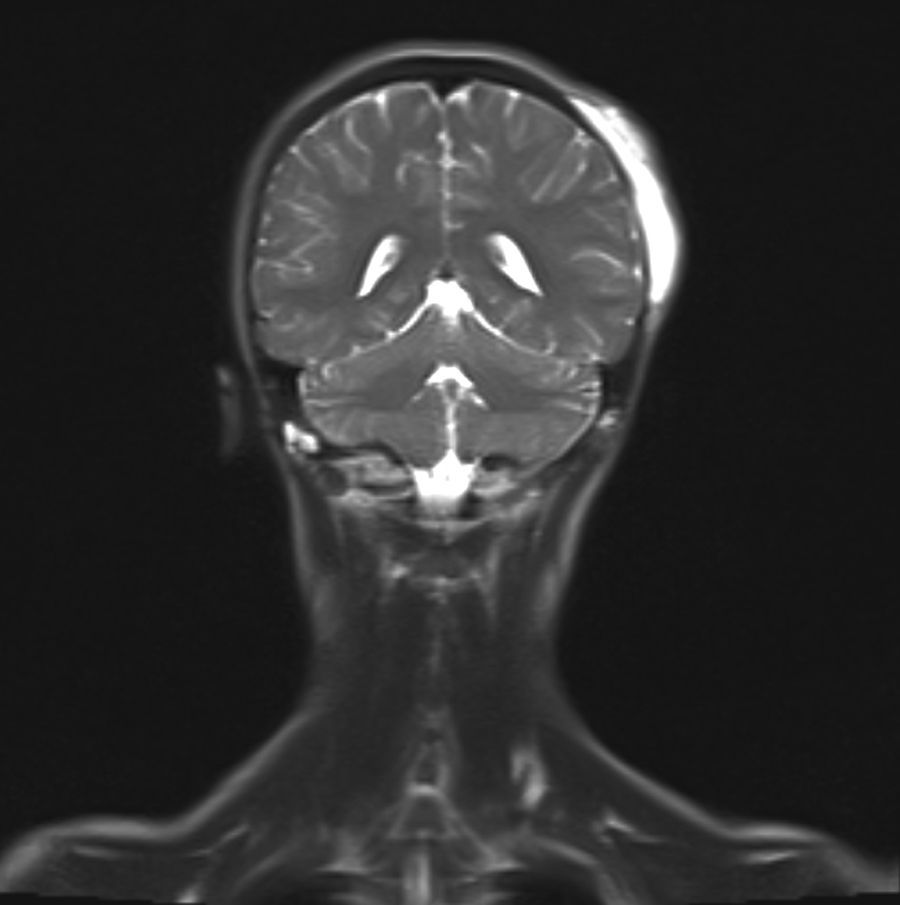
Se realiza estudio de anemia con evidencia de sangrado digestivo microscópico (sangre oculta en heces positiva), así como gammagrafía con eritrocitos marcados, en la que se observan depósitos focales de hematíes en abdomen. Ecografía abdominal normal. Se contacta con servicio de gastroenterología para estudio mediante endoscopia digestiva alta y colonoscopia bajo sedación profunda, en el que se observan varias lesiones polipoideas sésiles rojo-violáceas, de 15 a 25mm, de aspecto vascular, en segunda porción duodenal y en colon transverso (figs. 1 y 2). Se amplía estudio mediante RM craneal con contraste intravenoso y a nivel parietooccipital izquierdo se identifica una masa de unos 8cm, hiperintensa en T2 y que capta contraste, localizada en cuero cabelludo, tejido celular subcutáneo y que ocupa toda la tabla interna, externa y diploe craneal (fig. 3). Dicha masa tiene características de lesión vascular. No se identifica componente intracraneal ni alteración meníngea.



Se consulta el caso con dermatología que, ante los hallazgos encontrados, indica el diagnóstico de síndrome de Bean, que se confirma mediante biopsia: lesión vascular benigna que afecta desde la dermis superficial a la profunda, en cuya superficie se observa un patrón verrugoso con vasos dilatados, cavernosos, con contenido hemático, canales vasculares interconectados tipo linfangiomatoso, fenómenos de trombosis y recanalización vascular. Se inicia tratamiento con hierro oral y ácido fólico, así como profilaxis antituberculosa por Mantoux e IGRA positivos y se contacta con el centro de referencia de cirugía pediátrica para valoración de tratamiento quirúrgico.
La etiopatogenia del síndrome de Bean es desconocida. Se ha observado una asociación familiar con herencia autosómica dominante en algunos casos (mutación en gen del brazo corto del cromosoma 9), aunque la gran mayor parte de ellos son esporádicos. Afecta a todas las razas y a ambos sexos por igual. Las lesiones suelen estar presentes en la infancia y pueden permanecer estables o ir aumentando de tamaño con la edad (integrinas y semaforinas como factores neuroendocrinos implicados en su proliferación)1.
Las lesiones son malformaciones venosas que suelen ser pequeñas, de 1-2cm, azuladas o violáceas, circunscritas, de consistencia blanda y número variable2. Se localizan fundamentalmente en piel y tracto gastrointestinal (dentro de este, más frecuentemente en intestino delgado y colon izquierdo), aunque pueden encontrarse en cualquier órgano o tejido (sistema nervioso central, riñones, pulmones, hígado, hueso, ojos, etc.)3,4. Se suele manifestar con anemia ferropénica secundaria a sangrado digestivo crónico, aunque ocasionalmente puede presentarse en forma de sangrado agudo, vólvulo o invaginación. En otras localizaciones extradigestivas los síntomas estarán en relación con hemorragia o efecto compresivo a ese nivel (hemotórax, hemopericardio, paraparesia, epilepsia, etc.). A nivel hematológico también puede producir coagulopatía por hipofibrinogenemia y trombocitopenia de consumo1,5.
Su diagnóstico será fundamentalmente clínico-endoscópico, pudiendo utilizar pruebas radiológicas si existe sospecha de afectación en otras localizaciones. Se requiere un manejo multidisciplinario, con tratamiento inicial de la anemia mediante ferroterapia o transfusiones. Se han descrito casos en los que se ha intentado un manejo médico con resultados irregulares y escasa eficacia mediante agentes antiangiogénicos (corticoides, IFN), octeótrido, gammaglobulina intravenosa en altas dosis, danazol (antigonadotropinas) o anticonceptivos orales. Resulta más aceptado el tratamiento mixto endoscópico-quirúrgico, dirigido fundamentalmente a disminuir los requerimientos transfusionales en los casos graves, tratando las lesiones mediante diferentes técnicas, incluidas la escleroterapia6, la coagulación con argón plasma, la polipectomía con asa de diatermia7, la ligadura con bandas elásticas, resección y sutura. En muchas ocasiones el tratamiento endoscópico se realizará de forma intraoperatoria, tratando de ser globalmente lo más conservador posible, dada la frecuente recurrencia de las lesiones1,2,8.

