






El cáncer colorrectal es una de las neoplasias más frecuentes en los países occidentales y constituye un problema de salud pública en el ámbito mundial. En una proporción significativa de casos existe una agregación familiar de esta neoplasia, lo que se denomina cáncer colorrectal familiar para distinguirlo de las formas hereditarias bien establecidas. En la presente revisión se aporta el conocimiento actual referente a las causas genéticas subyacentes, las estrategias de cribado más adecuadas y las potenciales implicaciones pronósticas de esta entidad.
Colorectal cancer is one of the most common neoplasms in western countries and is a worldwide public health problem. A substantial number of cases show familial aggregation of the disease, termed familial colorectal cancer to distinguish it from well-established hereditary forms. The present review discusses current knowledge of the underlying genetic causes, the most appropriate screening strategies and the potential prognostic implications of this entity.
El cáncer colorrectal (CCR) es una de las neoplasias más frecuentes en los países occidentales y constituye un problema de salud pública en el ámbito mundial. En España, se estima que se diagnostican 26.000 nuevos casos cada año y casi el 50% de ellos fallece por la enfermedad1. A pesar de ser una entidad con un gran componente familiar, solamente una pequeña proporción de ellos (3–5%) corresponde a formas hereditarias con alta penetrancia, como son el síndrome de Lynch y las distintas poliposis colorrectales (poliposis adenomatosa familiar clásica y atenuada, síndrome de Peutz-Jeghers, poliposis juvenil, etc.)2. Cuando en el seno de una familia se diagnostican más casos de CCR de lo que correspondería por azar, es decir, existe una agregación familiar, se denomina «CCR familiar» y se distingue de los síndromes mencionados por no seguir un patrón de herencia tan bien definido. Según el estudio EPICOLON, el CCR familiar supone hasta el 30% de los casos de CCR en la población española3.
Los estudios de agregación familiar han contribuido a identificar las características que diferencian las formas familiares de las esporádicas, así como a avanzar en el conocimiento de su patogenia. Como en muchas otras neoplasias, en el desarrollo del CCR familiar intervienen tanto factores ambientales como genéticos. El componente ambiental desempeña un papel importante porque los miembros de una misma familia comparten hábitos dietéticos y estilos de vida parecidos. En este sentido, el consumo de carne roja y procesada, la obesidad, el alcohol, el tabaco así como la vida sedentaria se asocian a un mayor riesgo de CCR4. Sin embargo, la aportación de los factores ambientales parece menor que en otras neoplasias. Así, un estudio en población escandinava observó que el ambiente era el factor principal cuando se analizaban globalmente el conjunto de neoplasias, mientras que los factores genéticos representaban una contribución menor. No obstante, esta relación se invertía en las neoplasias de colon y próstata, y atribuían a la herencia un mayor efecto relativo5. Estos mismos resultados se obtuvieron en otro estudio más reciente que incluyó a todos los familiares de primer grado y consortes6.
Genética del cáncer colorrectal familiarEn la actualidad, está bien establecido que las formas hereditarias de CCR (poliposis colorrectales y síndrome de Lynch) están causadas por mutaciones en un único gen y siguen un patrón de herencia mendeliana. Corresponden a alelos poco frecuentes pero con una alta penetrancia, y su identificación se consiguió con éxito en las pasadas 2 décadas2. Sin embargo, en el CCR familiar los alelos implicados son más comunes y con una penetrancia menor, por lo que se postula que es la acción combinada de diversas variantes génicas la que contribuye al riesgo de desarrollar esta enfermedad. A este tipo de modelo se lo conoce como herencia poligénica e implica la interacción de múltiples variantes alélicas, cada una ligada a un riesgo bajo-moderado de CCR pero que en conjunto contribuyen al desarrollo de esta neoplasia7.
El avance en la identificación de los factores genéticos implicados en el CCR familiar procede, fundamentalmente, de estudios de asociación en los que se comparan casos afectados de esta neoplasia con controles sanos a fin de determinar la frecuencia de una o más variantes genómicas en ambos grupos y, así, establecer su implicación en la susceptibilidad para la enfermedad8.
Inicialmente, la mayoría de los estudios de asociación han evaluado la participación de determinados polimorfismos en genes implicados en la biología del CCR. En este sentido, está bien establecido que las mutaciones bialélicas en el gen MYH comportan el desarrollo de poliposis adenomatosa y CCR, con una penetrancia casi completa a los 60 años9, mientras que las mutaciones monoalélicas podrían estar implicadas en el riesgo familiar de tener esta neoplasia10. Por otro lado, el síndrome de Lynch es una enfermedad autosómica dominante asociada a mutaciones en los genes reparadores de los errores de replicación del ADN, principalmente MSH2, MLH1, MSH6 y PMS23. En este sentido, se ha postulado que la presencia de polimorfismos en estos genes podría asociarse al desarrollo de CCR familiar. Así, estudios de asociación han demostrado que el polimorfismo 415G>C (D132H) en el gen MLH1 incrementa el riesgo de tener CCR debido a una disminución en su función ATPasa11. Otro polimorfismo en el gen MLH1, la variante −93G>A, también se ha asociado a un riesgo aumentado de CCR (riesgo relativo [RR]: 1,84; intervalo de confianza [IC] del 95%: 1,20–2,83)12, mientras que el polimorfismo 116G>A (G39E) en el gen MSH6 se ha correlacionado con este incremento del riesgo sólo en hombres (RR: 1,27; IC del 95%: 1,04–1,54)13. Dado que las proteínas MYH y MSH6 intervienen juntas en el proceso de reparación del ADN, se ha propuesto que la combinación de variantes alélicas en ambos genes podría contribuir a un mayor riesgo de desarrollar CCR14. Finalmente, una variante común en el gen TGFBR1 (TGFBR1*6 A) también se ha asociado al riesgo de presentar diversos tipos de tumores, especialmente en homocigotos (RR: 2,02; IC del 95%: 1,18–3,48)15, mientras que la mutación I1307K del gen APC se ha asociado con una historia familiar de CCR en judíos Ashkenazi16.
Los estudios de asociación pangenómicos, ya aplicados a ciertas patologías, como la diabetes mellitus tipo 1, el infarto de miocardio o la enfermedad inflamatoria intestinal17, también se han utilizado en el campo del CCR. Estos estudios evalúan la implicación de múltiples single nucleotide polymorphism (SNP, polimorfismos de un solo nucleótido) distribuidos a lo largo de todo el genoma, los que se han identificado y ensamblado en bloques haplotípicos. Dentro de cada bloque, algunos SNP (tag SNP) representan la variación genética de un determinado gen o región genómica. De esta forma, los estudios de asociación reducen el número de SNP necesarios para una cobertura pangenómica y potencian el poder estadístico18.
Con estas aproximaciones pangenómicas, hasta el momento, se han identificado 10 variantes genómicas relacionadas con el CCR, que representan aproximadamente el 3% del exceso de riesgo familiar14. Una de ellas se halló en la región cromosómica 8q2419 y otra se localizó en el gen SMAD7 que actúa como antagonista intracelular de la señalización TGF-β20. Posteriormente, también se han implicado las regiones cromosómicas 15q13.321, 10p1422, 8q23.322 y 11q2323. Por último, un metaanálisis reciente ha identificado 4 loci de riesgo de CCR en las regiones 14q22.2, 16q22.1, 19q13.1 y 20p12.324,25. Aunque el riesgo individual asociado a cada SNP no es alto, su combinación en un modelo poligénico podría incrementarlo ya sea de forma aditiva o exponencial. Así, el riesgo aumentaría con el número de variantes alélicas (RR de cada alelo: 1,19; IC del 95%: 1,15–1,23) hasta 5 veces para individuos portadores de 7 o más variantes23. De esta manera, los portadores de múltiples alelos de riesgo podrían tener una probabilidad suficientemente alta de desarrollar CCR que justificara medidas de cribado específicas.
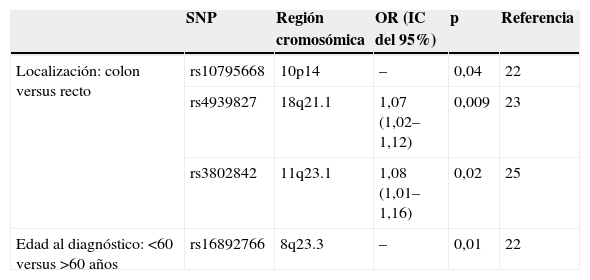
Con independencia del incremento del riesgo de CCR asociado a los alelos de susceptibilidad, es probable que éstos permitan clasificar a los pacientes en subgrupos con diferente presentación clínica, respuesta al tratamiento o pronóstico14. Así, se ha encontrado relación entre algunas variantes genéticas y ciertas características fenotípicas (tabla 1). Este aspecto se ha abordado de forma más exhaustiva en un estudio reciente realizado en el seno del proyecto EPICOLON, en el que se han correlacionado los 10 alelos de riesgo y sus correspondientes interacciones con las características personales y familiares de pacientes con CCR, mediante análisis de regresión logística. Este análisis ha permitido establecer que el alelo C en la región 8q23.3 (rs16892766) se asocia con un estadio tumoral más avanzado (OR: 1,48; IC del 95%: 1,15–1,90; p=2,5×10−3), que el alelo G en la región 8q24.21 (rs6983267) es más frecuente en pacientes con historia familiar de CCR (OR: 2,02; IC del 95%: 1,35–3,03; p=2,4×10−4) y que existe un efecto aditivo entre rs6983267 en la región 8q24.21 y rs9929218 en la región 16q22.2, asociado a la historia personal de adenoma colorrectal (portadores de ambos alelos minoritarios GG y AA, respectivamente, OR: 2,28; IC del 95%: 1,32–3,93; p=5,0×10–4) (manuscrito remitido para su evaluación). La demostración de una correlación entre las variantes genéticas y las características fenotípicas abre una nueva aproximación a la prevención del CCR mediante el diseño de estrategias, ajustada al riesgo de un determinado grupo de individuos o pacientes.
Correlación genotipo-fenotipo de polimorfismos de un solo nucleótido identificados en estudios de asociación pangenómicos
| SNP | Región cromosómica | OR (IC del 95%) | p | Referencia | |
| Localización: colon versus recto | rs10795668 | 10p14 | – | 0,04 | 22 |
| rs4939827 | 18q21.1 | 1,07 (1,02–1,12) | 0,009 | 23 | |
| rs3802842 | 11q23.1 | 1,08 (1,01–1,16) | 0,02 | 25 | |
| Edad al diagnóstico: <60 versus >60 años | rs16892766 | 8q23.3 | – | 0,01 | 22 |
IC: intervalo de confianza; OR: odds ratio; SNP: single nucleotide polymorphism ‘polimorfismos de un solo nucleótido’.
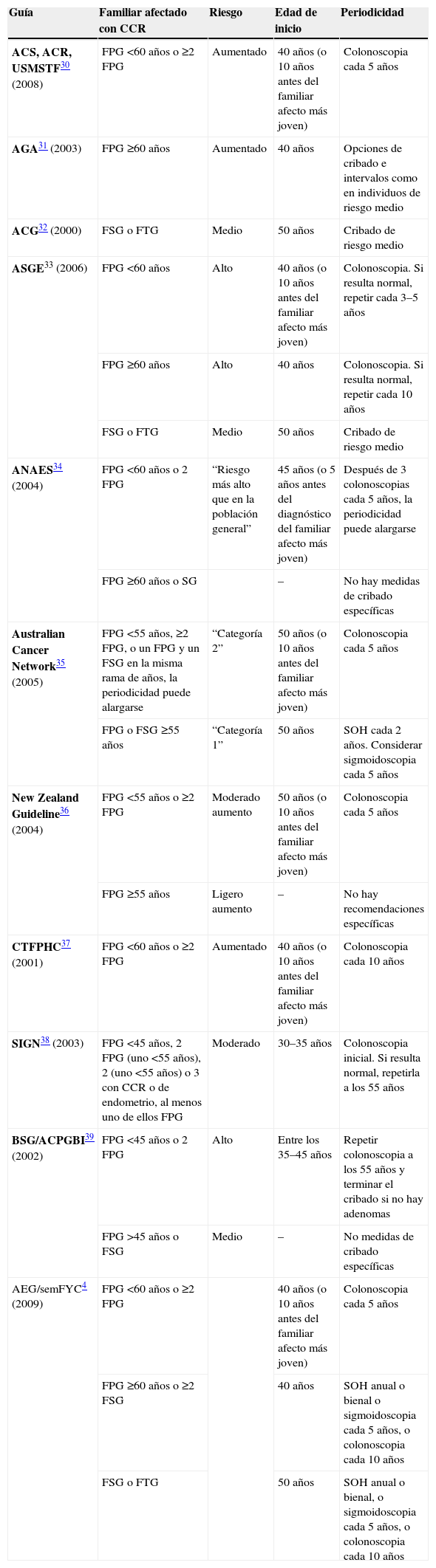
La ausencia de estudios prospectivos ha contribuido a que las guías de práctica clínica de prevención de CCR hayan establecido las recomendaciones en cribado en este grupo de riesgo de forma empírica (tabla 2). En general, todas las estrategias coinciden en establecer medidas de cribado más intensivas en los individuos con antecedentes familiares de CCR que las propuestas para la población de riesgo medio (individuos ≥50 años sin factores de riesgo personales ni familiares), ya sea con una edad de inicio del programa más temprana o con un acortamiento del intervalo entre exploraciones.
Estrategias de cribado en el cáncer colorrectal familiar
| Guía | Familiar afectado con CCR | Riesgo | Edad de inicio | Periodicidad |
| ACS, ACR, USMSTF30 (2008) | FPG <60 años o ≥2 FPG | Aumentado | 40 años (o 10 años antes del familiar afecto más joven) | Colonoscopia cada 5 años |
| AGA31 (2003) | FPG ≥60 años | Aumentado | 40 años | Opciones de cribado e intervalos como en individuos de riesgo medio |
| ACG32 (2000) | FSG o FTG | Medio | 50 años | Cribado de riesgo medio |
| ASGE33 (2006) | FPG <60 años | Alto | 40 años (o 10 años antes del familiar afecto más joven) | Colonoscopia. Si resulta normal, repetir cada 3–5 años |
| FPG ≥60 años | Alto | 40 años | Colonoscopia. Si resulta normal, repetir cada 10 años | |
| FSG o FTG | Medio | 50 años | Cribado de riesgo medio | |
| ANAES34 (2004) | FPG <60 años o 2 FPG | “Riesgo más alto que en la población general” | 45 años (o 5 años antes del diagnóstico del familiar afecto más joven) | Después de 3 colonoscopias cada 5 años, la periodicidad puede alargarse |
| FPG ≥60 años o SG | – | No hay medidas de cribado específicas | ||
| Australian Cancer Network35 (2005) | FPG <55 años, ≥2 FPG, o un FPG y un FSG en la misma rama de años, la periodicidad puede alargarse | “Categoría 2” | 50 años (o 10 años antes del familiar afecto más joven) | Colonoscopia cada 5 años |
| FPG o FSG ≥55 años | “Categoría 1” | 50 años | SOH cada 2 años. Considerar sigmoidoscopia cada 5 años | |
| New Zealand Guideline36 (2004) | FPG <55 años o ≥2 FPG | Moderado aumento | 50 años (o 10 años antes del familiar afecto más joven) | Colonoscopia cada 5 años |
| FPG ≥55 años | Ligero aumento | – | No hay recomendaciones específicas | |
| CTFPHC37 (2001) | FPG <60 años o ≥2 FPG | Aumentado | 40 años (o 10 años antes del familiar afecto más joven) | Colonoscopia cada 10 años |
| SIGN38 (2003) | FPG <45 años, 2 FPG (uno <55 años), 2 (uno <55 años) o 3 con CCR o de endometrio, al menos uno de ellos FPG | Moderado | 30–35 años | Colonoscopia inicial. Si resulta normal, repetirla a los 55 años |
| BSG/ACPGBI39 (2002) | FPG <45 años o 2 FPG | Alto | Entre los 35–45 años | Repetir colonoscopia a los 55 años y terminar el cribado si no hay adenomas |
| FPG >45 años o FSG | Medio | – | No medidas de cribado específicas | |
| AEG/semFYC4 (2009) | FPG <60 años o ≥2 FPG | 40 años (o 10 años antes del familiar afecto más joven) | Colonoscopia cada 5 años | |
| FPG ≥60 años o ≥2 FSG | 40 años | SOH anual o bienal o sigmoidoscopia cada 5 años, o colonoscopia cada 10 años | ||
| FSG o FTG | 50 años | SOH anual o bienal, o sigmoidoscopia cada 5 años, o colonoscopia cada 10 años |
ACG: American College of Gastroenterology; ACS: American Cancer Society; AEG: Asociación Española de Gastroenterología; AGA: American Gastroenterological Association; ANAES: Agence Nationale d’Accréditation et d’Évaluation en Santé; ASGE: American Society for Gastrointestinal Endoscopy; BSG/ACPGBI: British Society of Gastroenterology and the Association of Coloproctology of Great Britain and Ireland; CCR: cáncer colorrectal; CTFPHC: Canadian Task Force on Preventive Health Care; FPG: familiar de primer grado (hermanos, padres e hijos); FSG: familiar de segundo grado (abuelos, tíos y sobrinos); FTG: familiar de tercer grado (bisabuelos y primos); SemFYC: Sociedad Española de Medicina de Familia y Comunitaria; SIGN: Scottish Intercollegiate Guidelines Network; SOH: sangre oculta en heces; USMSTF: United States Multi-Society Task Force on Colorectal Cancer.
Existen múltiples estudios que demuestran un incremento del riesgo de tener CCR asociado a la presencia de antecedentes familiares de esta neoplasia. En un intento de aunar todos ellos y conseguir una mayor significación estadística se han realizado 3 metaanálisis con los estudios existentes. El primero de ellos analiza 27 estudios de casos y controles y de cohortes26, mientras que los otros 227,28 analizan 33 y 57 publicaciones, respectivamente. Es importante señalar que en el metaanálisis de Baglietto27 se realizan 2 aproximaciones: una que evalúa el riesgo de CCR desde la perspectiva de que el caso índice sea un familiar de un paciente afectado (RR tipo i), y otra en el que se estima el riesgo que confiere a sus familiares un paciente con CCR (RR tipo ii)27.
El riesgo de CCR a los 40 años de edad en un individuo con un familiar de primer grado (padres, hijos y hermanos) afectado es el mismo que el de la población general a los 50 años29. Por eso, la mayoría de las guías recomiendan, en el contexto de CCR familiar, iniciar el cribado a los 40 años, o 10 años antes de la edad al diagnóstico del familiar afectado más joven (lo que primero ocurra). Cuando el número de familiares de primer grado afectados es de 2 o más, el riesgo se multiplica por 426–28 (tabla 3), un aspecto que queda reflejado en todas las recomendaciones (tabla 2). Sin embargo, existe discrepancia en cuanto a las medidas e intervalos adoptados. Mientras que las guías americanas30–33, la francesa34, la australiana35, la neozelandesa36 y la española4 recomiendan colonoscopia cada 5 años, la canadiense lo alarga a 10 años37. Por otro lado, las guías escocesa38 y británica39 proponen una colonoscopia inicial y repetir sólo una más a los 55 años, y finalizar el cribado si ésta es normal (tabla 2).
Riesgo de cáncer colorrectal según el número de familiares de primer grado afectos
| Johns et al27 | Baglietto et al28 | Butterworth et al29 | |
| 1 FPG afectado | 2,25 (2,0–2,53) | 2,26 (1,86–2,73) | 2,24 (2,06–2,43) |
| 2 FPG afectados | 4,25 (3,01–6,08) | 3,95 (2,49–6,26) | 3,97 (2,60–6,06) |
| Uno o más hermanos afectados | 2,57 (2,19–3,02) | 2,52 (2,01–3,15) | 2,79 (2,36–3,29) |
| Uno o más progenitores afectados | 2,26 (1,87–2,72) | 2,15 (1,74–2,65) | 2,07 (1,83–2,34) |
FPG: familiar de primer grado (hermanos, padres e hijos).
Dentro de los familiares de primer grado afectados, los 3 metaanálisis muestran un riesgo mayor cuando éste es un hermano (RR: 2,57; IC del 95%: 2,52–2,79) que cuando lo es el padre (RR: 2,26; IC del 95%: 2,15–2,37), aunque sin alcanzar la significación estadística26–28 (tabla 3).
La probabilidad de desarrollar CCR es mayor cuanto más joven es el familiar afectado26–28, y aumenta 3 veces si el caso índice se diagnosticó entre los 45 y los 55 años de edad, y hasta 4 veces si lo fue antes de los 45 años40 (tabla 4). Las principales asociaciones americanas30–33 así como el grupo de trabajo español4 y el francés34 consideran que ante un paciente diagnosticado de CCR a una edad inferior a 60 años, el cribado en sus familiares debería comenzar a los 40 años. Otras guías, como la australiana35 o la neozelandesa36, reducen el punto de corte (edad del familiar diagnosticado de CCR) a los 55 años para indicar este cribado más precoz, mientras que en las guías británicas el punto de corte se sitúa en los 45 años38,39.
El riesgo de CCR también está incrementado cuando existen familiares de segundo (abuelos, tíos y sobrinos) y tercer grado (bisabuelos y primos) afectados, y se estima un RR de 1,73 (IC del 95%: 1,02–2,94)28. En estas situaciones, las medidas adoptadas por las diferentes guías varían. Mientras que las guías americanas30–33, australiana35 y española4 proponen las mismas medidas de cribado que en la población de riesgo medio, las guías francesa34 y británica39 consideran que tiene un riesgo más alto que la población general aunque no recomiendan unas medidas específicas de cribado. El resto de las guías no hacen referencia a este grupo de riesgo.
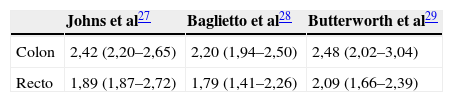
Finalmente, con respecto a la localización del tumor, los 3 metaanálisis coinciden en que el riesgo es mayor cuando el familiar afecto tiene un tumor localizado en el colon (RR: 2,42; IC del 95%: 2,20–2,48) que cuando éste se localiza en el recto (RR: 1,89; IC del 95%: 1,79–2,09)26–28 (tabla 5).
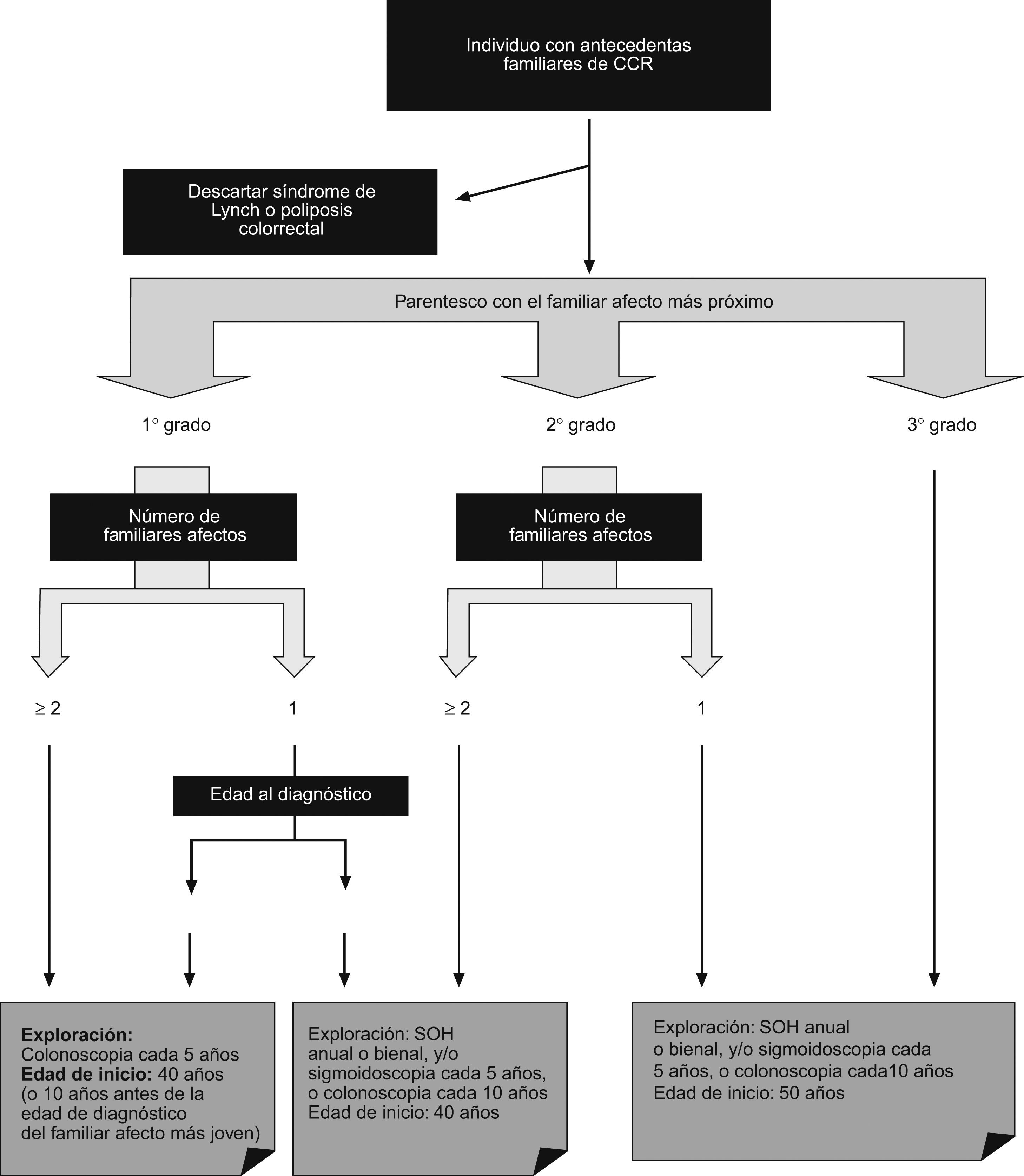
En definitiva, la diferencia entre las guías radica fundamentalmente en el carácter más restrictivo de unas frente a otras. La propuesta de la Asociación Española de Gastroenterología (AEG), la Sociedad Española de Medicina de Familia y Comunitaria (semFYC) y el Centro Cochrane Iberoamericano en su Guía de Práctica Clínica de Prevención del Cáncer Colorrectal (actualización del año 2009)4 queda reflejada en la figura 1.
.")
Estrategia de cribado en el cáncer colorrectal familiar (adaptado de la Guía de Práctica Clínica en Prevención del Cáncer Colorrectal4).
Es importante señalar, por último, que este incremento de riesgo familiar también se ha observado en relación con los adenomas. En un estudio de casos y controles se observó que entre los pacientes con CCR, el riesgo de tener un familiar con pólipos adenomatosos era de 1,5 (IC del 95%: 1,0–2,4), que aumentaba hasta 2,6 (IC del 95%: 1,3–5,1) si se consideraban sólo los adenomas avanzados (≥1cm o con componente velloso)41. Anteriormente, el National Polyp Study ya había estimado el riesgo de CCR en 2,59 (IC del 95%: 1,46–4,58) en hijos y padres de pacientes con adenomas diagnosticados antes de los 60 años de edad, y en 1,78 (IC del 95%: 1,8–2,67) cuando el diagnóstico se efectuó a una edad posterior42.
Pronóstico en el cáncer colorrectal familiarSi bien está plenamente establecido el incremento de riesgo de CCR cuando existen antecedentes familiares de esta enfermedad (véase apartado previo), pocos estudios han evaluado la influencia que tiene la historia familiar en el pronóstico de los pacientes con CCR.
Recientemente, ha aparecido un estudio que demuestra que en pacientes con CCR estadio iii tratado con cirugía y quimioterapia adyuvante, el antecedente de CCR en familiares de primer grado se asocia con una reducción significativa de la recurrencia y la mortalidad43. Así, comparado con los pacientes sin antecedentes familiares, se obtuvo una tasa ajustada entre los pacientes con más de un familiar de primer grado afectado de 0,72 (IC del 95%: 0,54–0,96) para la supervivencia libre de enfermedad; de 0,74 (IC del 95%: 0,55–0,99) para el riesgo de recurrencia, y de 0,75 (IC del 95%: 0,54–1,05) para la supervivencia global. Esta reducción en el riesgo de recurrencia o muerte aumenta con el número de familiares afectados, pero es independiente de la presencia de inestabilidad de microsatélites o de mutación en los genes reparadores de los errores de replicación del ADN43. Los resultados obtenidos en este estudio son consistentes con los observados en el registro japonés, que demostró una mejoría de la supervivencia a los 5 años en pacientes con antecedentes familiares de CCR, sin encontrar diferencias estadísticamente significativas en cuanto a la presencia de metástasis, el estadio clínico o la tasa de resección curativa44.
Los resultados de estos estudios apoyan la hipótesis de que en el CCR familiar existe un sustrato genético que podría influir no sólo en el riesgo de tener esta enfermedad, sino también en la supervivencia de estos pacientes45. Si estos resultados se confirmasen en estudios posteriores, la caracterización molecular de los pacientes con historia familiar de CCR podría llevar a la identificación de características genéticas predictivas de la respuesta al tratamiento14.
FinanciaciónEl CIBERehd (Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas) está financiado por el Instituto de Salud Carlos III.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Al Ministerio de Ciencia e Innovación (SAF 07-64873), a la Asociación Española contra el Cáncer (Fundación Científica y Junta de Barcelona) y a la Agència de Gestió d’Ajuts Universitaris i de Recerca (SGR 849).

