




Desde el descubrimiento del virus de la hepatitis C (VHC), se han desarrollado una plétora de modelos experimentales que nos han permitido el estudio del ciclo de vida del virus y la patogenia de las enfermedades hepáticas relacionadas con el VHC. Estos modelos van desde la inoculación de células en cultivo con suero de pacientes con hepatitis C hasta el uso de modelos «derivados» para el estudio de etapas concretas del ciclo de vida del VHC: las pseudopartículas retrovirales para el estudio de la entrada, los replicones para el estudio de la replicación y el modelo de cultivo celular capaz de reproducir todo el ciclo de vida completo (replicación y producción de partículas infecciosas). La utilización de estas herramientas ha sido y sigue siendo crucial para la identificación de posibles dianas terapéuticas en las diferentes etapas del ciclo de vida del virus y para el cribado de nuevos antivirales. Un claro ejemplo de ello es la reciente aprobación de dos inhibidores de la proteasa viral (boceprevir y telaprevir) en combinación con interferón pegilado (pegIFN) y ribavirina en la terapia contra la hepatitis crónica C. En esta revisión analizaremos los avances en la biología molecular del VHC y destacaremos posibles candidatos como nuevas dianas terapéuticas para el tratamiento de la infección por el VHC.
Since the discovery of the hepatitis C virus (HCV), a plethora of experimental models have evolved, allowing the virus's life cycle and the pathogenesis of associated liver diseases to be investigated. These models range from inoculation of cultured cells with serum from patients with hepatitis C to the use of surrogate models for the study of specific stages of the HCV life cycle: retroviral pseudoparticles for the study of HCV entry, replicons for the study of HCV replication, and the HCV cell culture model, which reproduces the entire life cycle (replication and production of infectious particles). The use of these tools has been and remains crucial to identify potential therapeutic targets in the different stages of the virus's life cycle and to screen new antiviral drugs. A clear example is the recent approval of two viral protease inhibitors (boceprevir and telaprevir) in combination with pegylated interferon and ribavirin for the treatment of chronic hepatitis C. This review analyzes the advances made in the molecular biology of HCV and highlights possible candidates as therapeutic targets for the treatment of HCV infection.
La infección por el virus de la hepatitis C (VHC) es la principal causa de hepatitis crónica, cirrosis y carcinoma hepatocelular, así como la indicación más frecuente de trasplante hepático en América, Europa y Japón. Según los datos más recientes de la Organización Mundial de la Salud (OMS), la hepatitis crónica C afecta aproximadamente a unos 150 millones de personas. Se estima que el VHC infecta cada año a 3-4 millones de personas y que anualmente mueren más de 350.000 personas por enfermedades hepáticas relacionadas con el VHC (WHO, Fact sheet No. 164, julio 2012).
Hasta hace pocos meses, la terapia estándar contra la hepatitis C se ha basado en la combinación de interferón alfa pegilado (pegIFN) y ribavirina. Dado que este tratamiento tiene una eficacia limitada, especialmente en los pacientes infectados por el VHC de genotipos 1 y 4, la reciente adición de inhibidores de la proteasa viral (telaprevir y boceprevir) a la terapia estándar con pegIFN y ribavirina ha supuesto un hito en la terapia contra el VHC, alcanzando una tasa de respuesta de hasta el 80% en pacientes infectados por el VHC de genotipo 11–3. Sin embargo, este tratamiento no es igualmente efectivo contra todos los genotipos del VHC ni en todas las cohortes de pacientes, por lo que urge el desarrollo de fármacos de nueva generación que, solos o en combinación, sean capaces de eliminar el virus, contrarrestando la variabilidad genética de este, la aparición de mutantes de resistencia y los mecanismos de defensa del huésped.
Las investigaciones sobre la biología molecular del VHC y su interacción con el huésped se han visto limitadas durante años por la falta de un modelo animal apropiado y un sistema de cultivo celular in vitro capaz de reproducir el ciclo replicativo completo del VHC, incluida la producción de partículas virales infecciosas4–6. Además, por razones desconocidas, el VHC derivado del suero de pacientes replica en un nivel muy bajo y de forma muy poco reproducible en cultivos primarios de hepatocitos humanos y en líneas celulares derivadas de hepatoma7–9. En esta revisión discutiremos de forma práctica diversos aspectos de la biología molecular del VHC y cómo el desarrollo de modelos experimentales in vitro nos ha permitido no solo el estudio del ciclo de vida del VHC y su interacción con su huésped, sino también la identificación de nuevas dianas terapéuticas para el tratamiento de la infección por el VHC.
Clasificación y variabilidad genéticaEl VHC pertenece al género hepacivirus de la familia Flaviviridae10. Además del género hepacivirus, esta familia contiene virus de los géneros pestivirus (por ejemplo, virus de la diarrea bovina) y flavivirus (por ejemplo, virus del dengue y de la fiebre amarilla) y otros virus todavía sin clasificar, designados GBV-A y GBV-C11,12. Aunque tradicionalmente el VHC ha sido considerado como el único miembro del género hepacivirus, en 2005 se incluyó el virus GBV-B, un virus hepatotropo que infecta tamarinos11,13. Recientemente, Kapoor et al. han identificado un homólogo canino del VHC en muestras respiratorias de perros14. El hepacivirus canino es el primer hepacivirus aislado de un animal no primate y filogenéticamente más cercano al VHC descrito hasta la fecha. Además, posteriormente se ha detectado la presencia de virus similares al hepacivirus canino en caballos y se han caracterizado 8 nuevos hepacivirus genéticamente diversos15.
La variabilidad genética es una de las características biológicas más relevantes del VHC. Mediante el análisis comparativo de las secuencias genómicas de virus aislados en diferentes zonas geográficas se han podido identificar hasta 7 genotipos distintos del VHC, denominados del 1 al 7 según el orden de descubrimiento. Los distintos genotipos difieren entre sí en un 30-35% de los nucleótidos (nt). Asimismo, cada uno de estos genotipos está dividido en subtipos, los cuales difieren entre sí en un 20-25% de la secuencia de nt y son identificados mediante letras, por ejemplo, 1a, 1b, 2a, 2b, 3a, etc.16.
El VHC se diversifica también a nivel de cuasiespecie17. Este término se refiere a la diversidad que existe entre los virus que circulan dentro de un mismo individuo. La diversidad de secuencias se genera continuamente durante la replicación viral debido a que la polimerasa viral no tiene capacidad correctora de errores (1 error cada 104 o 105 nt), y a que la tasa de producción de nuevos viriones es muy alta (1012 viriones por día) resultando en un gran tamaño poblacional18,19.
Estructura del virus de la hepatitis CEl VHC es un virus pequeño (∼ 50nm de diámetro) y con envuelta. Los primeros análisis por microscopia electrónica de partículas virales aisladas de muestras de suero e hígado de pacientes y chimpancés infectados mostraron la presencia de partículas esféricas de unos 33-70nm20–23. Posteriormente, con el desarrollo del sistema de cultivo celular del VHC4–6, se han podido visualizar 2 tipos de partículas virales por inmunoelectromicroscopia: partículas de 30-35nm de diámetro correspondientes a las nucleocápsides y partículas de 50-60nm, que probablemente sean los viriones infecciosos5.
Por analogía con otros miembros de la familia Flaviviridae, se cree que el VHC podría adoptar una estructura icosaédrica. Sin embargo, datos recientes sugieren que el VHC tiene una estructura pleomórfica, con las glucoproteínas E1 y E2 incrustadas en la membrana lipídica derivada del retículo endoplasmático de la célula huésped24,25. Bajo la membrana lipídica se encuentra la nucleocápside, compuesta por múltiples copias de la proteína core rodeando al ARN genómico. Las partículas virales aisladas de plasma muestran diferentes densidades y se cree que esto se debe a su asociación con complejos de inmunoglobulinas o con lipoproteínas celulares de baja densidad26–28.
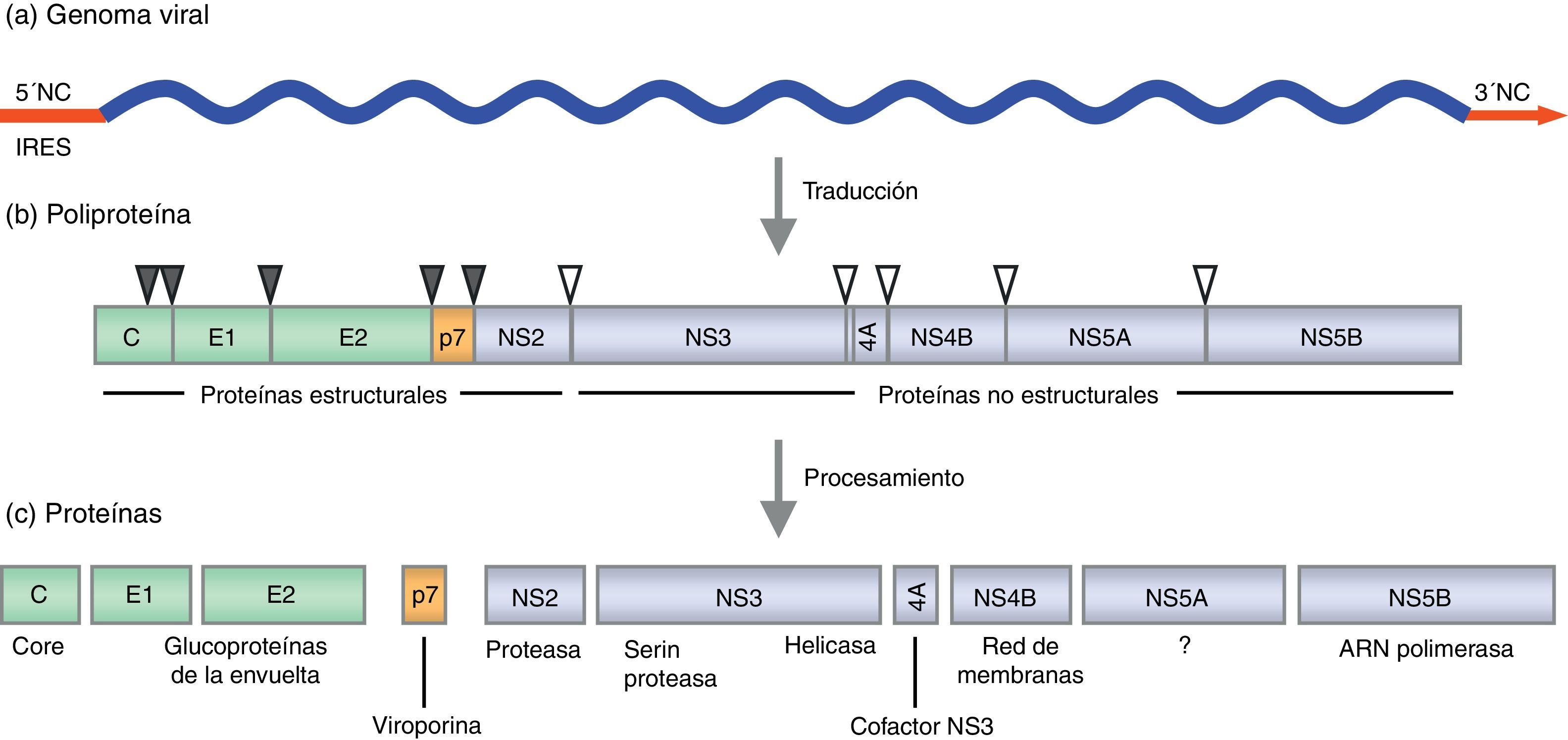
Organización genómicaEl genoma del VHC está constituido por una única molécula de ARN, de polaridad positiva. El genoma completo contiene aproximadamente 9.600 nt y consta de un único marco de lectura de entre 9.024 y 9.111 nt, dependiendo del genotipo. El marco de lectura está flanqueado por 2 regiones no traducidas en los extremos 5’ y 3’, las cuales tienen una conservada estructura secundaria (fig. 1). Estas 2 regiones del virus son esenciales para la replicación y la traducción del ARN. El extremo 5’ (341 nt) actúa como sitio interno de entrada al ribosoma (IRES, internal ribosome entry site), desde el cual se puede iniciar la síntesis proteica. También contiene señales de la replicación esenciales para la síntesis de los intermediarios replicativos de ARN de polaridad negativa, que servirán como moldes para la replicación viral. Recientemente, se ha descrito que el micro-ARN-122 (miRNA-122, específico del hígado) se une al extremo 5’, promoviendo la replicación y traducción del virus29–31. Además, el secuestro de miRNA-122 mediante oligonucleótidos antisentido, reduce la replicación del VHC tanto en cultivo celular como in vivo, lo que sugiere que el miRNA-122 podría ser una diana para la terapia antiviral32,33. El extremo 3’ es esencial para la replicación34. Contiene aproximadamente 225 nt y consta de 3 partes: una primera región de longitud variable de unos 30-40 nt, una cola poli-U/UC también de longitud variable y una secuencia terminal de 98 nt muy conservada denominada 3’ X35.
 Representación lineal del genoma viral: el marco de lectura abierto (azul) y las regiones no traducidas 5’ y 3’ (rojo). b) Organización de las proteínas en el precursor poliproteico: proteínas estructurales (verde) y no estructurales (lila). Los triángulos grises círculos en negro indican los sitios de corte de las peptidasas celulares para dar lugar a las proteínas estructurales. Los triángulos blancos se refieren a los sitios de corte de las proteasas virales para dar lugar a las proteínas no estructurales. c) Proteínas virales y su función.")
Esquema de la organización genómica del VHC y las proteínas virales. a) Representación lineal del genoma viral: el marco de lectura abierto (azul) y las regiones no traducidas 5’ y 3’ (rojo). b) Organización de las proteínas en el precursor poliproteico: proteínas estructurales (verde) y no estructurales (lila). Los triángulos grises círculos en negro indican los sitios de corte de las peptidasas celulares para dar lugar a las proteínas estructurales. Los triángulos blancos se refieren a los sitios de corte de las proteasas virales para dar lugar a las proteínas no estructurales. c) Proteínas virales y su función.
La traducción del marco de lectura da lugar a una poliproteína precursora de unos 3.000 aminoácidos (aa), aproximadamente. Esta poliproteína es procesada tanto por proteasas virales como del huésped dando lugar a 3 proteínas estructurales (core, E1, E2), la proteína p7 y 6 proteínas no estructurales (NS2, NS3, NS4A, NS4B, NS5A y NS5B) (fig. 1).
La proteína core se encuentra formando parte de la nucleocápside. Además, se ha señalado que core interacciona directamente con diversas proteínas y vías de señalización celulares que podrían ser importantes para el ciclo de vida del VHC (por ejemplo, apoptosis, transcripción de genes, metabolismo lipídico, respuesta inmunitaria, etc.)36. Las 2 glucoproteínas de la envuelta, E1 y E2, son componentes esenciales de la envuelta del virión y son las responsables de la entrada y fusión viral37,38. La proteína p7, una proteína integral de membrana de función todavía poco conocida. Estudios in vitro sugieren que p7 pertenece a la familia de las viroporinas, y que podría actuar como canal iónico de calcio39–41. Asimismo, estudios recientes demuestran que esta proteína está implicada en la morfogénesis y la secreción de partículas infecciosas42.
Las 6 proteínas restantes son proteínas no estructurales cuya función se centra básicamente en el procesamiento de la poliproteína y en la replicación viral. La proteína NS2 junto con el dominio aminoterminal de NS3 forma la proteasa NS2-NS3, una proteasa dependiente de cinc que procesa el lugar de corte entre NS2 y NS3. Además, NS2 interacciona molecularmente con p7 y E2 y tiene un papel crucial en la producción de viriones43–47. La proteína NS3 contiene un dominio serina proteasa en el extremo aminoterminal y un dominio helicasa/ATPasa en el extremo carboxiterminal. La NS4A es un cofactor que se asocia a la proteasa NS348. La proteasa NS3-NS4A es esencial para el ciclo de vida del VHC, catalizando el procesamiento del resto de las proteínas no estructurales en los sitios NS3/NS4A, NS4A/NS4B, NS4B/NS5A y NS5A/NS5B. Por este motivo, la proteasa NS3-NS4A es una de las dianas virales más interesantes en la terapia contra el VHC.
La NS4B es una proteína integral de membrana altamente hidrofóbica, en la que se predicen 4 dominios transmembrana y un extremo N-terminal implicado en la unión a las membranas intracelulares49,50. Su principal función es alterar las membranas intracelulares y formar una red de membranas tipo «andamio» denominada membranous web que es esencial para la formación del complejo de replicación51–53. La NS5A es una metaloproteasa dependiente de cinc muy fosforilada, cuya función en el ciclo de vida del VHC no está claramente definida54. Sin embargo, diversos estudios demuestran que, además de desempeñar un papel importante en la replicación viral55–57 y en la producción de partículas infecciosas58–60, interacciona con un gran número de proteínas celulares, regulando diversas vías de señalización61–64. La NS5B es la polimerasa viral. Es una ARN polimerasa dependiente de ARN (RdRp), que se ha demostrado que es capaz de sintetizar ARN de novo y dependiente de cebador (primer). Tiene la estructura típica de las polimerasas (estructura de mano derecha), con los dominios dedos, palma y pulgar. La interacción entre los dominios dedos y pulgar sitúa el centro catalítico en la base de la palma, haciendo posible la síntesis del ARN de cadena positiva y negativa del VHC65,66.
Además de las 10 proteínas codificadas en el marco de lectura abierto, se ha descrito la existencia de una forma diferente de la proteína core originada a partir del desplazamiento a través de desplazamientos del marco de lectura del ribosoma. La proteína F (frameshift) o ARFP (alternate reading frame protein) es una proteína quimérica que contiene los aminoácidos de los extremos N-terminal y C-terminal de core y una nueva parte central codificada por el marco de lectura +1. Esta proteína se expresa de manera natural durante la infección por el VHC de genotipo 1a, ya que se han encontrado anticuerpos antiproteína F67.
Ciclo de vida del virus de la hepatitis CEl VHC replica preferentemente en el citoplasma de los hepatocitos, aunque también se ha descrito la presencia del VHC en otros compartimentos o tipos celulares como células mononucleares de sangre periférica68,69, células dendríticas70 o sistema nervioso central71.
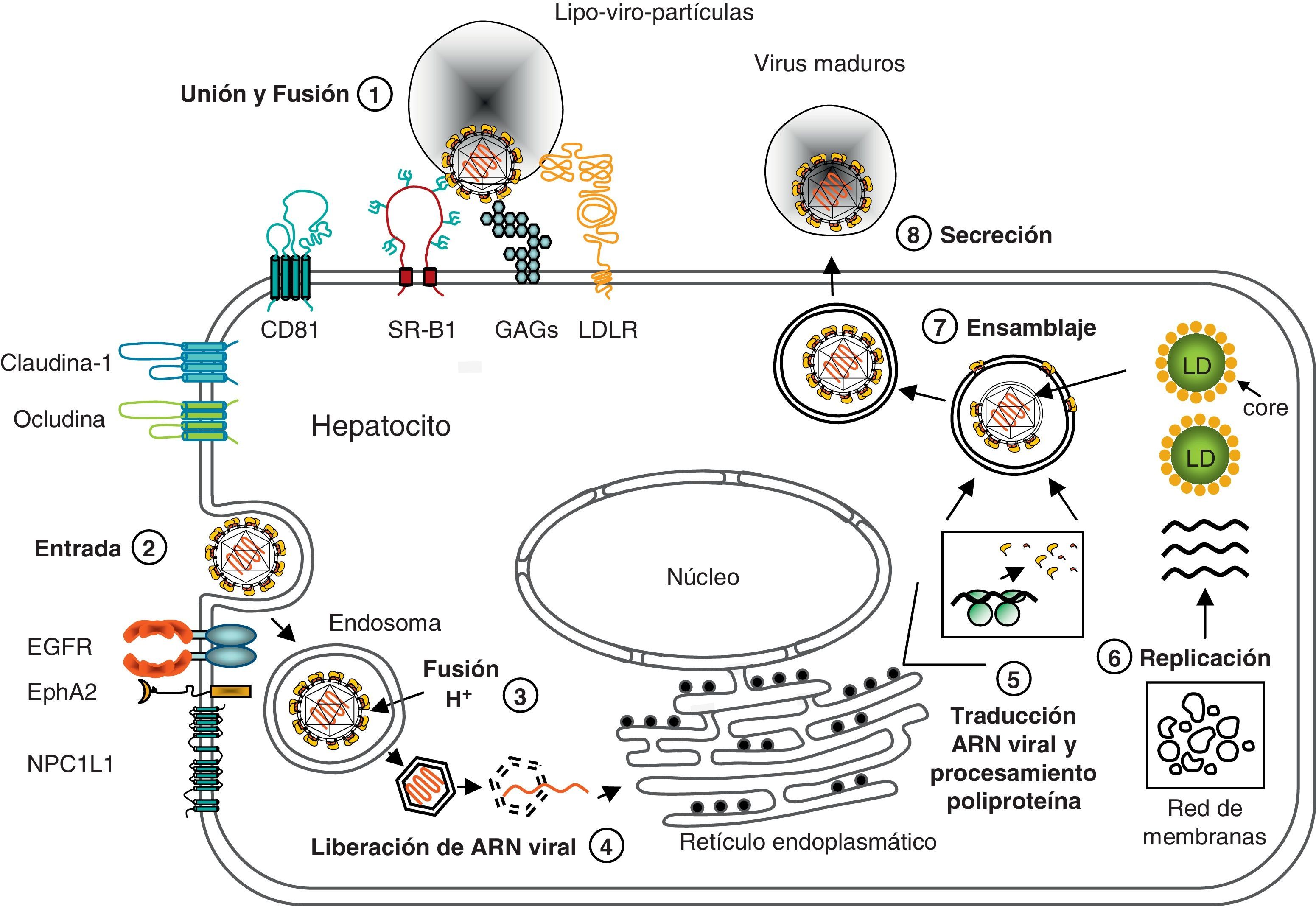
La partícula viral circula libre o asociada a lipoproteínas72. El mecanismo exacto mediante el cual el VHC alcanza el citoplasma e inicia la replicación no se conoce con precisión. Las evidencias acumuladas hasta la fecha indican que para llegar desde el endotelio sinusoidal hasta el espacio de Disse, el VHC asociado a lipoproteínas puede o bien difundir a través de las fenestras de las células endoteliales o bien ser capturado y «transmitido» a los hepatocitos adyacentes por las lectinas de tipo C DC-SIGN (dendritic cell-specific ICAM-3 grabbing non-integrin) y L-SIGN (liver/lymph node cell-specific ICAM-3 grabbing non-integrin), expresadas mayoritariamente en células dendríticas y en el endotelio sinusoidal, respectivamente73,74. La interacción inicial del virión con la membrana celular está mediada por glucosaminoglucanos (GAG) como el heparán sulfato, que ayudarían a concentrar el virus en la superficie celular75–77 (fig. 2). Como consecuencia de la asociación entre el VHC y las lipoproteínas, también se ha propuesto que el receptor de LDL (LDLR) puede participar en la entrada del VHC en los hepatocitos78. A continuación, el VHC interacciona de manera específica y secuencial con el SR-B1 (scanvenger receptor B1)79, la tetraspanina CD8180 y las proteínas de las uniones estrechas (tight junction proteins), claudina-181 y ocludina82.
. 1) La interacción inicial del virión con la membrana celular está mediada por glucosaminoglucanos (GAG). A continuación, el VHC interacciona de manera secuencial con los receptores LDLR, SR-B1, CD81, claudina-1 y ocludina. Los receptores EGFR y EphA2 (y probablemente NPCL1L1) regularían la asociación entre CD81 y claudina-1 y la fusión de la envuelta viral con la membrana plasmática. 2) La entrada en el hepatocito se produce por endocitosis dependiente de clatrina. 3) La fusión de la envuelta del virus con la membrana del endosoma es un proceso mediado por la acidificación del endosoma. 4) Desencapsidación y liberación del ARN viral. 5) Traducción del ARN viral y procesamiento de la proliproteína por proteasas virales y del huésped. 6) Replicación del ARN viral en el complejo de replicación, asociado a la red de membranas. 7) Formación de la cápside y ensamblaje de los nuevos viriones alrededor de los LD. 8) Las partículas del VHC se asocian a lipoproteínas y son liberadas a través de la vía de secreción celular.")
Representación gráfica del ciclo de vida del VHC. La partícula viral circula libre o asociada a lipoproteínas (lipo-viro-partículas). 1) La interacción inicial del virión con la membrana celular está mediada por glucosaminoglucanos (GAG). A continuación, el VHC interacciona de manera secuencial con los receptores LDLR, SR-B1, CD81, claudina-1 y ocludina. Los receptores EGFR y EphA2 (y probablemente NPCL1L1) regularían la asociación entre CD81 y claudina-1 y la fusión de la envuelta viral con la membrana plasmática. 2) La entrada en el hepatocito se produce por endocitosis dependiente de clatrina. 3) La fusión de la envuelta del virus con la membrana del endosoma es un proceso mediado por la acidificación del endosoma. 4) Desencapsidación y liberación del ARN viral. 5) Traducción del ARN viral y procesamiento de la proliproteína por proteasas virales y del huésped. 6) Replicación del ARN viral en el complejo de replicación, asociado a la red de membranas. 7) Formación de la cápside y ensamblaje de los nuevos viriones alrededor de los LD. 8) Las partículas del VHC se asocian a lipoproteínas y son liberadas a través de la vía de secreción celular.
Recientemente se han identificado 2 receptores tirosina cinasa, EGFR (epidermal growth factor receptor) y EphA2 (ephrin type-A receptor 2) como cofactores de la entrada del VHC. Estos receptores regulan la asociación entre CD81 y claudina-1 y la fusión de la envuelta viral con la membrana plasmática83. Asimismo, en otro estudio muy reciente se ha demostrado que el receptor NPC1L1 (Niemann-Pick C1-like 1), implicado en la absorción intestinal de colesterol, también tiene un papel en la entrada del VHC, probablemente en la fusión de las membranas viral y celular84.
Tras la unión a sus receptores, el VHC entra en los hepatocitos por endocitosis dependiente de clatrina85. La fusión de la envuelta del virus con la membrana del endosoma tiene lugar en los endosomas tempranos y es un proceso mediado por el pH ácido de los endosomas86. Después de la desencapsidación, el ARN viral es liberado al citoplasma y, dirigido por el IRES de la región 5’, se traduce directamente dando lugar a una única proteína que es procesada por proteasas celulares y virales. Las proteínas estructurales (core, E1 y E2) y la proteína p7 son procesadas por la peptidasa señal del retículo endoplasmático, mientras que las proteínas no estructurales son procesadas por 2 proteasas virales, la proteasa NS2-3 y la serina proteasa NS3-4A.
Las proteínas virales forman el complejo de replicación, un complejo multiproteico asociado a membranas intracelulares52,87. Esta red de membranas se localiza muy próxima a la membrana del retículo endoplasmático, sugiriendo que deriva de este compartimento. Dentro de este complejo, el ARN de polaridad positiva es copiado a un ARN complementario de polaridad negativa, que sirve de molde para la síntesis de la progenie de ARN genómico de polaridad positiva. Estos ARN genómicos servirán para la síntesis de nuevos ARN complementarios negativos, para la traducción o serán encapsidados. El modelo actual de ensamblaje de las partículas virales postula que las nucleocápsides formadas en el citoplasma adquieren la envuelta de glucoproteínas mientras brotan a través del retículo endoplasmático. Una vez ensambladas, las partículas del VHC son liberadas a través de la vía de secreción celular. Aunque estos procesos son bastante desconocidos, se ha propuesto que existe una asociación entre el metabolismo de los lípidos y los procesos de ensamblaje y liberación del virus72,88. Estudios recientes han demostrado que el core se localiza principalmente en la membrana que rodea las gotas lipídicas (LD, lipid droplets) y que esta asociación es importante para la formación de partículas infecciosas in vitro. De este modo, el core reclutaría las proteínas no estructurales y los complejos de replicación en sitios adyacentes a las gotas lipídicas, donde se ensamblarían los viriones89,90. Asimismo, la vía de síntesis de VLDL está implicada en la secreción del VHC. El uso de inhibidores o de ARN de interferencia contra componentes de la vía de síntesis de VLDL, como por ejemplo la proteína de transferencia de triglicéridos microsomales, ApoB y ApoE, reprimen la secreción de partículas infecciosas91–95.
Replicación in vitro del virus de la hepatitis C derivado del suero de pacientesDesde el descubrimiento de VHC en 1989, muchos investigadores han intentado infectar células en cultivo con virus aislado de suero de pacientes infectados por el VHC, utilizando tanto líneas celulares humanas de origen hepático como de otros orígenes y/o especies. En un primer estudio sobre la replicación del VHC in vitro, Shimizu et al. inocularon células MOLT-4 (línea celular humana derivada de células T) con un suero que contenía VHC96. Siete días después de la inoculación, no solo se detectó la presencia de ARN de cadena negativa en las células, sino también la expresión de los antígenos virales de la cápside y NS4 por inmunofluorescencia, demostrando así que estas células eran susceptibles a la replicación del VHC. Sin embargo, el nivel de replicación del VHC en estas células fue muy bajo y el sistema resultó infructuoso para su aplicación in vitro. El análisis de otras líneas de origen linfoide llevó a la identificación de un clon de la línea de celular HPB-Ma (derivada de células T), capaz de soportar la replicación del VHC de forma consistente97. Además, los autores demostraron que existía una correlación entre la infectividad de diversos inóculos del VHC in vivo (chimpancés) y su infectividad in vitro. Si bien diversos estudios han descrito la susceptibilidad de líneas celulares derivadas de células T, B y hepatocitos fetales humanos a la infección por el VHC después de su inoculación con suero de pacientes98–103, el mantenimiento y propagación del virus en estos sistemas de cultivo es bastante complejo, debido probablemente a los bajos niveles de replicación del VHC in vitro.
El tropismo hepático del VHC se demostró por primera vez en estudios in vitro cuando 6 de 7 sueros de pacientes positivos para el VHC infectaron preferentemente a las células PH5CH (línea celular humana derivada de hepatocitos) que a la células MT-2 (línea celular humana derivada de células T)104. A partir de entonces, los investigadores concentraron sus esfuerzos en la inoculación de líneas celulares de origen hepático: se establecieron sistemas de cultivo a largo plazo utilizando líneas celulares derivadas de hepatoma humano (Huh7, HepG2)8 y se consiguió infectar cultivos primarios de hepatocitos humanos con suero de pacientes positivos para el VHC9. A pesar de todo, los modelos basados en la inoculación de cultivos primarios o diferentes líneas celulares con sueros de pacientes con hepatitis C presentaron una baja reproducibilidad y su aplicabilidad se limitó a determinadas muestras de suero muy seleccionadas. Por ello, estos sistemas prácticamente se abandonaron con el descubrimiento de los replicones subgenómicos de genotipos 1a105 y 1b55,106, que replican de manera autónoma y, preferentemente, en los subclones seleccionados de la línea celular de hepatoma humano Huh7, como por ejemplo, Huh7.5107 o Huh7-Lunet108.
Modelos experimentales para el estudio del virus de la hepatitis C in vitroEntrada: pseudopartículas viralesDurante los años 1990, debido a la falta de un sistema de cultivo celular para la propagación del VHC, se utilizaron sistemas celulares clásicos de expresión transitoria o estable para estudiar la función de las proteínas del VHC. Para el estudio de la entrada del VHC en sus células diana, en el año 2003, Bartosch et al.109, e independientemente Hsu et al.110, desarrollaron el sistema de infección de las pseudopartículas del VHC (VHCpp). Ambos grupos aprovecharon la propiedad de los retrovirus de que pueden incorporar glucoproteínas heterólogas en su envuelta, en este caso concreto las glucoproteínas de la envuelta E1 y E2 del VHC.
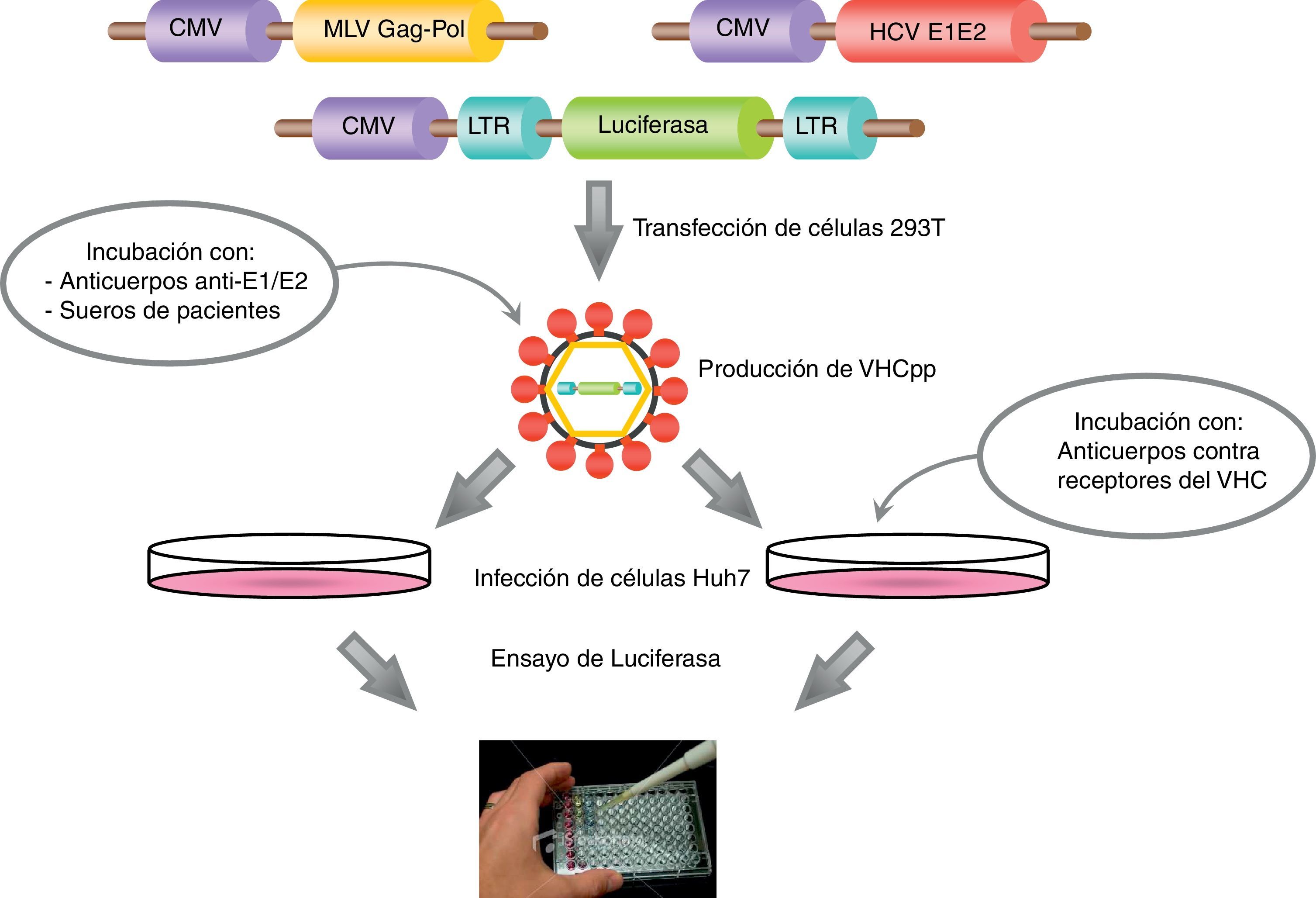
Las VHCpp se producen mediante la cotransfección de células 293T (células embrionarias de riñón humano) con vectores de expresión que codifican para: a) las glucoproteínas del VHC; b) las proteínas retrovirales (cápside y polimerasa) y la señal de empaquetamiento, y c) un gen reportero (luciferasa o proteína verde fluorescente, GFP). Los vectores retrovirales utilizados para la producción de las VHCpp están basados en el virus de la leucemia murina (MLV) o en el virus de la inmunodeficiencia humana (VIH-1). Las VHCpp son secretadas al medio de cultivo y a partir de las 48h después de la transfección, se recogen los sobrenadantes y se utilizan para infectar las células diana, como por ejemplo, Huh7 y sus derivadas (fig. 3). La infectividad de las VHCpp ha sido analizada en diversas líneas celulares y en cultivos primarios de hepatocitos humanos. Curiosamente, las VHCpp presentan un tropismo claro por las líneas celulares de origen hepático y por los hepatocitos humanos, mientras que, en general, en las líneas celulares que no son de origen hepático o bien no se infectan o bien la infección es mínima109.
. Las VHCpp secretadas al medio de cultivo pueden utilizarse para infectar células diana (por ejemplo, Huh7, Huh7.5, etc.). Asimismo, también es posible realizar ensayos de neutralización bien preincubando las VHCpp con anticuerpos anti-E1/E2 o sueros de pacientes infectados, o bien preincubando las células diana con anticuerpos contra los receptores de la entrada del VHC.")
Diagrama de la producción de VHCpp mediante la cotransfección de células 293T con 3 plásmidos que contienen los genes que codifican para las glucoproteínas de la envuelta del VHC, la cápside retroviral y la señal de empaquetamiento, y un gen reportero (luciferasa). Las VHCpp secretadas al medio de cultivo pueden utilizarse para infectar células diana (por ejemplo, Huh7, Huh7.5, etc.). Asimismo, también es posible realizar ensayos de neutralización bien preincubando las VHCpp con anticuerpos anti-E1/E2 o sueros de pacientes infectados, o bien preincubando las células diana con anticuerpos contra los receptores de la entrada del VHC.
Cabe destacar que el sistema VHCpp sirve únicamente para el estudio de la entrada del VHC en sus células diana. Aun así, se ha utilizado con éxito para el estudio de los procesos inmediatos a la unión del virus a la célula y en la identificación de los receptores clave en la entrada viral, revelando que la entrada del VHC en los hepatocitos es un proceso muy complejo y muy regulado111.
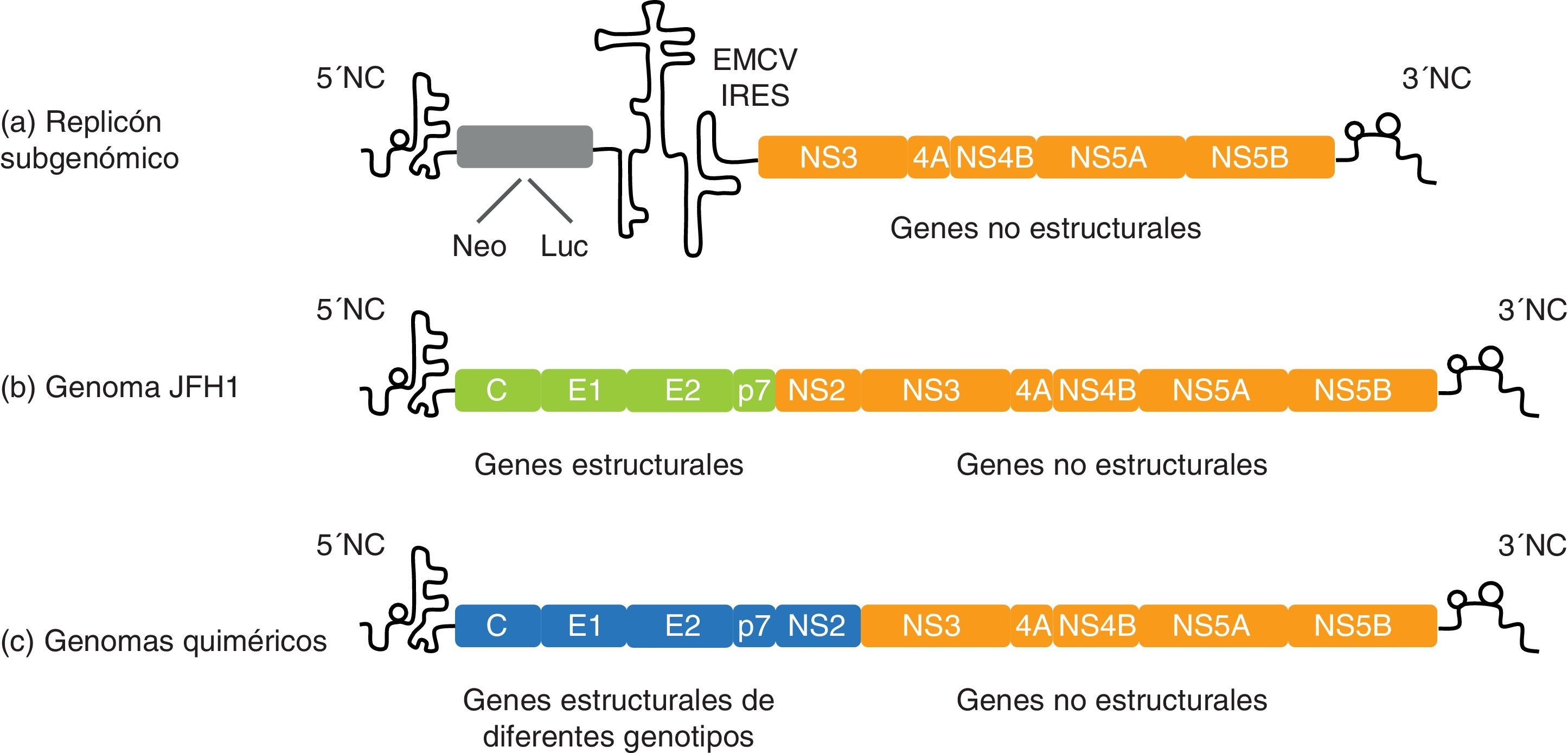
Replicación: replicón subgenómicoEn el año 1999 se produjo un gran avance en el campo de la biología molecular del VHC cuando Lohmann et al. desarrollaron los primeros replicones subgenómicos funcionales del VHC106. Por definición, los replicones son moléculas de ácidos nucleicos capaces de autoduplicarse. En caso del VHC, los replicones de primera generación fueron derivados de un genoma del VHC de genotipo 1b (denominado Con 1) en el que se reemplazaron los genes estructurales por un gen de resistencia a la neomicina (neomicina fosfotransferasa). También se incluyó un segundo elemento IRES para aumentar la traducción de las proteínas no estructurales del VHC, que constituyen la maquinaria de replicación del virus (fig. 4). Así pues, estas construcciones subgenómicas y bicistrónicas pueden autorreplicarse, a través de la síntesis de una cadena negativa, y propagarse en cultivo celular durante años56,106. Las líneas celulares más permisivas para ello son Huh7.5 y Huh7-Lunet107.
 Ejemplo de un replicón subgenómico bicistrónico compuesto por: el extremo 5’ no codificante (NC) dirigiendo la traducción del gen marcador o de selección, el IRES del virus de la encefalomiocarditis (EMCV), los genes de la replicasa del VHC y el extremo 3’NC. b) Modelo de cultivo celular del VHC: para la producción de partículas virales se necesitan las proteínas no estructurales y los extremos 5’ y 3’ de JFH1. c) Los genes core, E1, E2 y p7 se pueden sustituir por los de otras cepas del VHC para construir genomas quiméricos y estudiar in vitro las propiedades de los diferentes genotipos del VHC.")
Representación esquemática del replicón subgenómico del VHC y cultivo celular del VHC basado en JFH1. a) Ejemplo de un replicón subgenómico bicistrónico compuesto por: el extremo 5’ no codificante (NC) dirigiendo la traducción del gen marcador o de selección, el IRES del virus de la encefalomiocarditis (EMCV), los genes de la replicasa del VHC y el extremo 3’NC. b) Modelo de cultivo celular del VHC: para la producción de partículas virales se necesitan las proteínas no estructurales y los extremos 5’ y 3’ de JFH1. c) Los genes core, E1, E2 y p7 se pueden sustituir por los de otras cepas del VHC para construir genomas quiméricos y estudiar in vitro las propiedades de los diferentes genotipos del VHC.
Dado que el sistema del replicón subgenómico del VHC no contiene los genes estructurales, no es capaz de producir partículas virales. Por ello, también se desarrollaron replicones que contenían todo el genoma completo del VHC y un marcador de selección112. Estos replicones genómicos necesitaban mutaciones adaptativas al cultivo celular para replicar de forma eficiente y en niveles suficientemente altos para la selección por el gen marcador. Aunque los replicones genómicos del VHC fueron muy prometedores, la aparición de mutaciones adaptativas parecía interferir con la producción de partículas113.
Los replicones subgenómicos han sido herramientas muy valiosas en los estudios de los mecanismos de replicación del VHC y en la caracterización bioquímica y estructural del complejos de replicación viral52,114 y en el ensayo de nuevos fármacos contra la hepatitis C (inhibidores de la proteasa y la polimerasa del VHC), incluida la identificación de resistencias a los antivirales115–117.
Ciclo de vida completo: modelo de cultivo celular del virus de la hepatitis CEn 2003, Kato et al. descubrieron un replicón subgenómico basado en una secuencia de genotipo 2a que, por razones desconocidas, replicaba a niveles entre 50 y 500 veces mayores en comparación con los replicones basados en Con 1, sin la necesidad de mutaciones adaptativas118. Este replicón se construyó a partir de un genoma del VHC de genotipo 2a denominado JFH 1 (Japanese fulminant hepatitis), obtenido de un paciente japonés que sufrió una hepatitis fulminante119. Dos años más tarde, 3 grupos independientes desarrollaron el modelo del cultivo celular del VHC (VHCcc) basado en el genoma completo de JFH 1 y que era capaz de replicar y producir partículas infecciosas in vitro4–6 (fig. 4). Concretamente, la transfección de células Huh7 (o sus derivadas) con ARN de JFH 1 conlleva la producción y liberación de partículas virales al medio de cultivo, que son infecciosas en cultivo celular, en chimpancé y en ratón5,6.
Una mejora importante del sistema ha sido la generación de virus quiméricos basados en JFH 1 con gran capacidad replicativa. Básicamente, estos genomas quiméricos contienen los extremos 3’ y 5’ y la región NS3-NS5B de JFH 1 (esencial para que se produzca replicación y producción de partículas virales), y la región core-NS2 procedente de otra cepa de genotipo 2a denominada J66,120. La quimera J6/JFH1 replica en cultivo celular y produce títulos virales unas 100 veces más elevados que su genoma parental JFH1. Además de esta quimera intragenotípica, también se han generado virus quiméricos con todos los genotipos del VHC. A diferencia de la quimera J6/JFH1, estas quimeras intergenotípicas necesitan mutaciones adaptativas al cultivo celular120–123. Dado que no existe ningún otro genoma del VHC capaz de reproducir el ciclo de vida completo del virus con la misma eficiencia que JFH 1 y que el genotipo del VHC influye en la respuesta al tratamiento antiviral, el hecho de disponer de genomas quiméricos de los distintos genotipos del VHC es especialmente relevante para desarrollo de nuevas estrategias antivirales, la evaluación de vacunas y la caracterización de la respuesta inmunitaria humoral en una amplia variedad de genotipos.
Otras estrategias con el fin de optimizar el modelo del VHCcc han sido la selección de clones de células especialmente permisivos a la replicación del virus4,6, el estudio sistemático de mutaciones adaptativas al cultivo celular, especialmente la identificación aquellas que aumentan los valores virales sin afectar a la replicación 32, y la generación de VHCcc que contienen genes reporteros (por ejemplo, la luciferasa o la proteína fluorescente verde) para facilitar la detección de la infección76,124.
Dianas terapéuticas en las diferentes etapas del ciclo de vida del virus de la hepatitis CLa entrada viral como diana terapéuticaTal y como se ha descrito en el apartado 4, el VHC necesita diversos receptores con diferente localización en la membrana celular. Esto implica que la entrada del VHC es un proceso complejo y sumamente regulado, pero que a su vez ofrece un gran número de posibles dianas terapéuticas. Potencialmente, la entrada viral se podría inhibir tanto mediante el bloqueo de la partícula viral como de los factores celulares de la entrada. La infección por el VHC se puede impedir mediante el uso de anticuerpos neutralizantes contra las proteínas E1 y/o E2 de la envuelta del virus125. Estos anticuerpos neutralizantes han demostrado ser muy potentes al neutralizar cepas de diferentes genotipos in vitro126,127 pero su eficacia in vivo es mucho menor128,129. Esto se podría deberse por un lado a la gran variabilidad genética del VHC, particularmente en las glucoproteínas de la envuelta, por lo que se requerirían anticuerpos neutralizantes de amplio espectro. Por otro lado, el virus circula asociado a lipoproteínas que podrían enmascarar los epítopos neutralizantes. Finalmente, el VHC también puede infectar a las células vecinas mediante transmisión célula a célula, escapando así de la neutralización por los anticuerpos neutralizantes.
El bloqueo de los receptores de la entrada del VHC CD81, SR-B1 y claudina-1ha demostrado ser eficaz tanto in vitro como in vivo (ratón), independientemente del genotipo del VHC130–133. El uso de inhibidores de la entrada tiene especial interés para impedir la recurrencia de la hepatitis C en pacientes que han recibido un trasplante hepático. Actualmente, se está realizando un estudio piloto de fase clínica 1 para evaluar la seguridad y eficacia preliminar del antagonista de SR-B1 ITX 5061 en pacientes infectados por el VHC y que han recibido un trasplante hepático. Por otro lado, la identificación de EGFR, EphA2 y NPC1L1 como factores de la entrada del virus abre nuevas oportunidades para el desarrollo de nuevos inhibidores. Un aspecto interesante es que actualmente existen 2 moléculas comercializadas, erlotinib (inhibidor de EGFR) y ezetimibe (inhibidor de NPC1L1), que han demostrado actividad antiviral in vitro e in vivo (ratón)83,84.
La replicación viral como diana terapéuticaLa replicación del VHC puede ser inhibida de forma eficiente con inhibidores de la proteasa NS3/4A. En 2011 se aprobaron 2 inhibidores de la proteasa viral, boceprevir y telaprevir, para el tratamiento de la infección por el VHC en combinación con pegIFN y ribavirina. Con la triple terapia, la respuesta virológica sostenida ha aumentado de forma significativa en pacientes infectados por el VHC de genotipo 1. Sin embargo, este tratamiento es menos efectivo contra los genotipos no 1 y conlleva la aparición de efectos secundarios graves1,134. Probablemente, los inhibidores de proteasa de segunda generación vayan dirigidos a disminuir los efectos adversos y ampliar su espectro de eficacia.
La replicación del VHC también puede ser inhibida con inhibidores de la polimerasa viral NS5B, que es la responsable de la replicación viral. Existen 2 tipos de inhibidores de la polimerasa: los análogos de nucleósidos y los no nucleósidos. En general, los análogos de nucleósidos son activos contra múltiples genotipos debido a que se unen al sitio activo de la polimerasa y este está muy conservado entre genotipos. Por el contrario, los inhibidores no nucleósidos son inhibidores alostéricos, por lo que aparentemente son más dependientes del genotipo viral135. Recientemente se han reportado datos clínicos muy prometedores del análogo de nucleósido sofosbuvir (GS-7977). En los ensayos clínicos de fase 2, este fármaco fue muy efectivo contra todos los genotipos del VHC, al ser administrado tanto con como sin pegIFN136–138. Actualmente, se encuentra en fase 3 de desarrollo clínico en pacientes con hepatitis crónica C y fase 2 para la prevención de la recurrencia de la hepatitis C en pacientes que han recibido un trasplante hepático.
La proteína no estructural NS4B es esencial para la replicación viral y la formación de los complejos de replicación. Aunque se desconoce el mecanismo exacto por el cual ejerce su función, NS4B también podría representar una posible diana terapéutica contra la infección por el VHC. En 2008, Einav et al. demostraron que NS4B era capaz de unirse al ARN del VHC a través del extremo 3’ de la cadena negativa y que el clemizol hidrocloruro (antihistamínico clásico) era un inhibidor potente de esta unión139. Asimismo, mediante estudios funcionales utilizando el replicón subgenómico se demostró que, aunque el clemizol inhibía la replicación de forma modesta, ejercía un efecto sinérgico al combinarse con los inhibidores de la proteasa telaprevir o boceprevir140. En 2010 se completó un estudio clínico de fase 1b en el que se evaluó la seguridad y tolerabilidad de clemizol en pacientes con hepatitis C que no habían sido tratados previamente. Sin embargo, los resultados de este ensayo clínico todavía no han sido publicados.
La proteína NS5A tiene un papel importante en la replicación viral y en la formación de partículas infecciosas. El primer inhibidor de NS5A, daclatasvir, ha demostrado ser un antiviral potente in vitro e in vivo, siendo capaz de incrementar la actividad antiviral del pegIFN y la ribavirina141,142. Actualmente, se están realizando ensayos clínicos en los que se combinan daclatasvir con asunaprevir (inhibidor de la proteasa NS3) en un régimen libre de pegIFN.
Durante el ciclo viral intervienen numerosos factores celulares que también podrían ser buenos candidatos para la terapia antiviral, solos o en combinación con pegIFN y ribavirina o con los antivirales directos. Las ciclofilinas son proteínas celulares con actividad cis-trans isomerasa (facilita el plegamiento de las proteínas), que se expresan en muchos tejidos humanos y están implicadas en numerosos procesos fisiológicos y en la replicación del VHC. Alisporivir es el prototipo de una nueva clase de inhibidores de la ciclofinas: es un análogo de la ciclosporina A exento de efecto inmunosupresor porque no interacciona con la calcineurina. Estudios in vitro han demostrado que alisporivir inhibe la replicación del VHC impidiendo la interacción entre la ciclofilina A y la proteína NS5A de VHC143,144. La eficacia de alisporivir también ha sido demostrada in vivo145 y aunque se encontraba en fases avanzadas de desarrollo clínico para el tratamiento de pacientes con hepatitis crónica C, la FDA ha decidido aplazar el ensayo por un tema de seguridad.
Otra alternativa interesante como diana antiviral es el miRNA-122. Como se ha comentado anteriormente, el miRNA-122 es específico del hígado y se une al extremo 5’ del genoma del VHC, promoviendo la replicación y traducción del virus29,122. Estudios in vitro han demostrado que el oligonucleótido antisentido miravirsen es un antagonista potente contra los genotipos el 1 al 6 del VHC32. Además, la administración de miravirsen a chimpancés infectados por el VHC causó una caída drástica de la carga viral de forma persistente, sin que se evidenciara la aparición de resistencias ni efectos adversos graves (excepto un descenso notable de los niveles de colesterol en suero)33. Recientemente se han publicado los resultados del estudio clínico de prueba de concepto en el que se ha demostrado la seguridad y eficacia de miravirsen en pacientes infectados por el VHC de genotipo 1146. Esta prevista la realización de más ensayos clínicos con miravirsen solo o en combinación con antivirales directos como por ejemplo el telaprevir.
La formación de partículas infecciosas como diana terapéuticaEl ensamblaje y la liberación de las partículas virales son los procesos menos conocidos del ciclo de vida del VHC. Se sabe que este proceso está muy relacionado con el metabolismo lipídico y que tanto las proteínas estructurales como las no estructurales interaccionan con esta vía a distintos niveles147. Por ello, cabría la posibilidad de bloquear la formación de partículas virales con antivirales contra las proteínas del virus o contra factores celulares como, por ejemplo, determinados componentes del metabolismo de los lípidos.
La proteína core ofrece varias ventajas como posible diana terapéutica. Por un lado, es esencial para la formación de la nucleocápside viral y, en consecuencia, para la formación de las partículas infecciosas. Por otro lado, está muy conservada entre los genotipos del VHC y es la que presenta la menor variabilidad genética de las proteínas virales en pacientes con hepatitis crónica C. La oligomerización del core es un paso necesario para la formación de la nucleocápside viral, por lo que su inhibición impediría la formación de partículas infecciosas. Estudios recientes han reportado la identificación de péptidos y moléculas pequeñas capaces de inhibir, no solo la dimerización del core, sino también la producción de virus en cultivo celular148–150. La proteína p7 podría ser otra diana interesante. La pérdida de p7 suprime completamente la producción de partículas infecciosas, y la pérdida de la su actividad como canal iónico interfiere con la producción de virus en cultivo celular40,42,43. Sin embargo, los antagonistas de los canales iónicos investigados hasta la fecha han demostrado un efecto antiviral muy modesto151.
El plegamiento correcto de las proteínas de la envuelta E1 y E2 es importante tanto para la entrada como para la formación de partículas infecciosas. Dado que E1 y E2 son proteínas muy glucosiladas, se desarrollaron inhibidores de la α-glucosidasas como posibles antivirales en la etapa de ensamblaje del virión. Dos inhibidores de la α-glucosidasa, UT-231B (un iminoazúcar) y celgosivir (profármaco de la castanoespemina) llegaron a fase clínica 2, pero ninguno de los 2 progresó a fases más avanzadas del desarrollo clínico: UT-231B por baja eficacia y celgosivir sin justificar el motivo152. En un estudio reciente se ha demostrado que DGAT1 (diacylglycerol O-acyltransferase 1) es un factor esencial para la localización de core alrededor de los lipid droplets153, por lo que quizás también podría ser un buen candidato como diana terapéutica. Por otro lado, dado que la morfogénesis de las partículas virales está estrechamente ligada a la biogénesis de VLDL, factores celulares asociados a esta vía como ApoB, ApoE y MTP (microsomal triglyceride transfer protein) podrían ser posibles dianas terapéuticas. Por ejemplo, el flavonoide cítrico naringenina es capaz de inhibir la secreción de VLDL in vitro e in vivo y la producción de virus en cultivo celular. La naringenina inhibe la secreción de ApoB mediante la inhibición de la actividad de MTP y la transcripción de las enzimas HMGR (3-hydroxy-3-methyl-glutaryl-coenzyme A reductase) y ACAT2 (acyl-coenzyme A: cholesterol acyltransferase 2) también implicadas en el metabolismo lipídico154. Asimismo, diversos estudios in vitro han indicado la capacidad de las estatinas para inhibir la replicación del VHC155. Sin embargo, los resultados de los estudios realizados en pacientes con hepatitis C son contradictorios, por lo que su aplicabilidad en humanos queda en entredicho156–158.
Conclusiones y perspectivasLos avances en el conocimiento de las diferentes etapas del ciclo de vida del VHC han permitido que el desarrollo de terapias contra la infección por el VHC aumente de forma exponencial durante los últimos años. La terapia triple estándar actual (pegIFN, ribavirina y telaprevir/boceprevir) ha mejorado sustancialmente la tasa de respuesta de los pacientes infectados por el VHC de genotipo 1. Sin embargo, este tratamiento no es siempre efectivo y a menudo está contraindicado debido a la aparición de efectos adversos en múltiples poblaciones de pacientes. El descubrimiento de nuevas dianas terapéuticas abrirá nuevas líneas de desarrollo de medicamentos más potentes, capaces de curar a la mayoría, sino a todos, los pacientes con hepatitis crónica C. En este sentido tiene especial relevancia el desarrollo de fármacos seguros dirigidos a tratar cohortes especiales de pacientes como pacientes que han recibido un trasplante hepático, pacientes coinfectados por otros virus (VHB o VIH) y/o pacientes con manifestaciones extrahepáticas habitualmente secundarias a reacciones autoinmunes, depósito generalizado de inmunocomplejos y trastornos linfoproliferativos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

