




El carcinoma sarcomatoide de intestino delgado es un tumor extremadamente raro cuyas manifestaciones clínicas son insidiosas e inespecíficas, las cuales abarcan desde tumor abdominal difuso, hasta hemorragia gastrointestinal u obstrucción intestinal. Estos datos explican el retraso importante del diagnóstico definitivo, la poca eficacia del tratamiento y el pronóstico reservado. Hasta el momento existen solamente 20 casos publicados. En el presente trabajo se presenta un caso de carcinoma sarcomatoide con localización yeyunal, y se subrayan las peculiaridades clínico-patológicas, a la vez que se discute una asociación hipotética con la enfermedad autoinmunitaria el síndrome de Sjögren.
Sarcomatoid carcinoma is an extremely rare small bowel tumor whose clinical manifestations are insidious and nonspecific, ranging from diffuse abdominal pain to gastrointestinal bleeding or intestinal occlusion. Thus, diagnostic delay is highly common with poor treatment outcome and prognosis. To date, only 20 cases have been reported in the literature. We describe the case of a small bowel sarcomatoid carcinoma localized in the jejunum, with emphasis on the clinical and pathological features of this entity. The hypothetical association with Sjögren's syndrome, an autoimmune disease, is also discussed.
El carcinoma primario de intestino delgado es un tumor extremadamente raro, que alcanza una incidencia de 1-15/100.000 habitantes/año1, y que representa un 2% del total de tumores gastrointestinales2 y menos del 0,4% de todos los tumores malignos3. Entre las posibles causas de la incidencia baja de neoplasias a ese nivel, se encuentran: la existencia de un tránsito rápido de contenido líquido intestinal, que evita un contacto prolongado con la mucosa; la presencia de una carga bacteriana menos importante que la del colon, y la presencia de tejido linfoideo con secreción importante de inmunoglobulina (Ig) A, que desempeña un papel protector importante.
La mayoría de los carcinomas primarios de intestino delgado son adenocarcinoma (45%), carcinoide (29%), linfoma maligno (16%) y sarcoma (10%)4.
Observación clínicaPresentamos el caso de un varón de 80 años, diagnosticado previamente de hipertensión arterial, artritis reumatoide con síndrome de Sjögren de larga evolución, hernia hiatal, antecedentes de trombosis venosa profunda, glaucoma con atrofia óptica, portador de prótesis de cadera, y que sigue tratamiento habitual con mirtazapina, rabeprazol, prednisona 5mg y metoclopramida.
El paciente es hospitalizado por cuadro de dolor en el epigastrio, de 5 meses de evolución, de carácter sordo, continuo, sin relación con ciclo digestivo, acompañado de vómitos frecuentes, biliosos, oscuros, malolientes, diarrea, pérdida de unos 17kg de peso y anorexia. Fue estudiado previamente por la misma clínica en otro centro hospitalario, en el que se le diagnosticó colelitiasis no complicada, hernia hiatal, diverticulosis en sigma y hemorroides internas.
En el examen del paciente destacaba postración, coloración cetrina, estado nauseoso, ausencia de fiebre y una presión arterial 100/60mmHg. Tenía una «ptisis bulbi» del ojo izquierdo. El examen pulmonar era normal y en la exploración cardíaca destacaba arritmia cardíaca sin soplos. El abdomen era blando, depresible, no distendido, con escasos movimientos peristálticos, sin signos de irritación peritoneal, ni agrandamiento de órganos internos. Las extremidades eran normales, sin datos de artritis ni trombosis venosa profunda.
Analíticamente destacaba una insuficiencia renal leve (creatinina 1,6mg/dl, urea 55mg/dl) con hiponatremia (sodio 134mEq/l, potasio 4,1mEq/l, cloro 97mEq/l), hipoalbuminemia (3g/dl) y ascenso de la β2-microglobulina (8,5mg/dl), con normalidad en función hepática, metabolismo del hierro y otros marcadores tumorales como antígeno carcinoembrionario (CEA), antígeno prostático específico, marcador tumoral 19,9 y alfafetoproteína. En el estudio hematológico destacaba una anemia normocítica normocrómica (hemoglobina 8,8g/dl, volumen corpuscular medio 83fl, CHM 32g/dl) y una velocidad de sedimentación globular de 47mm, con normalidad de los otros parámetros.
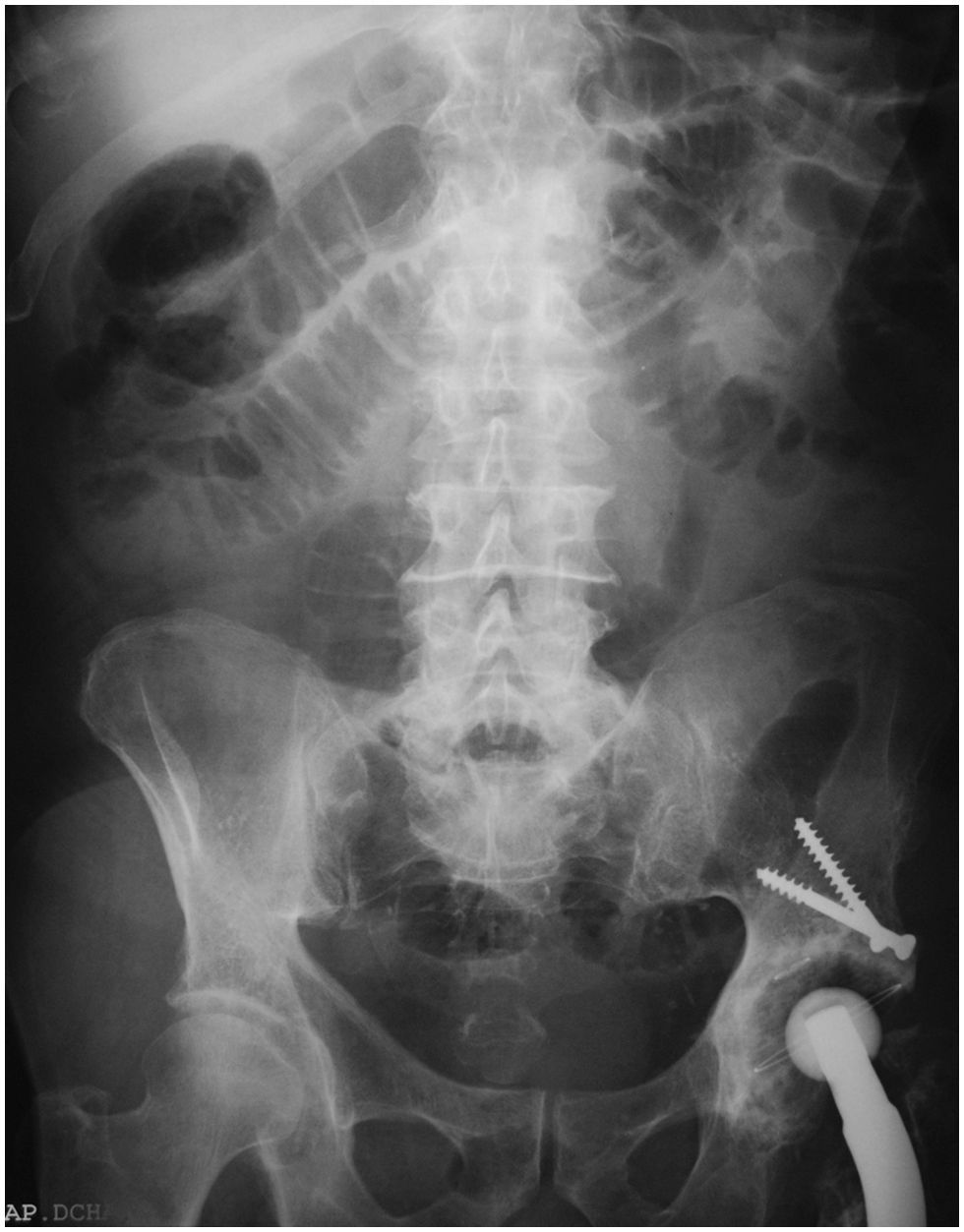
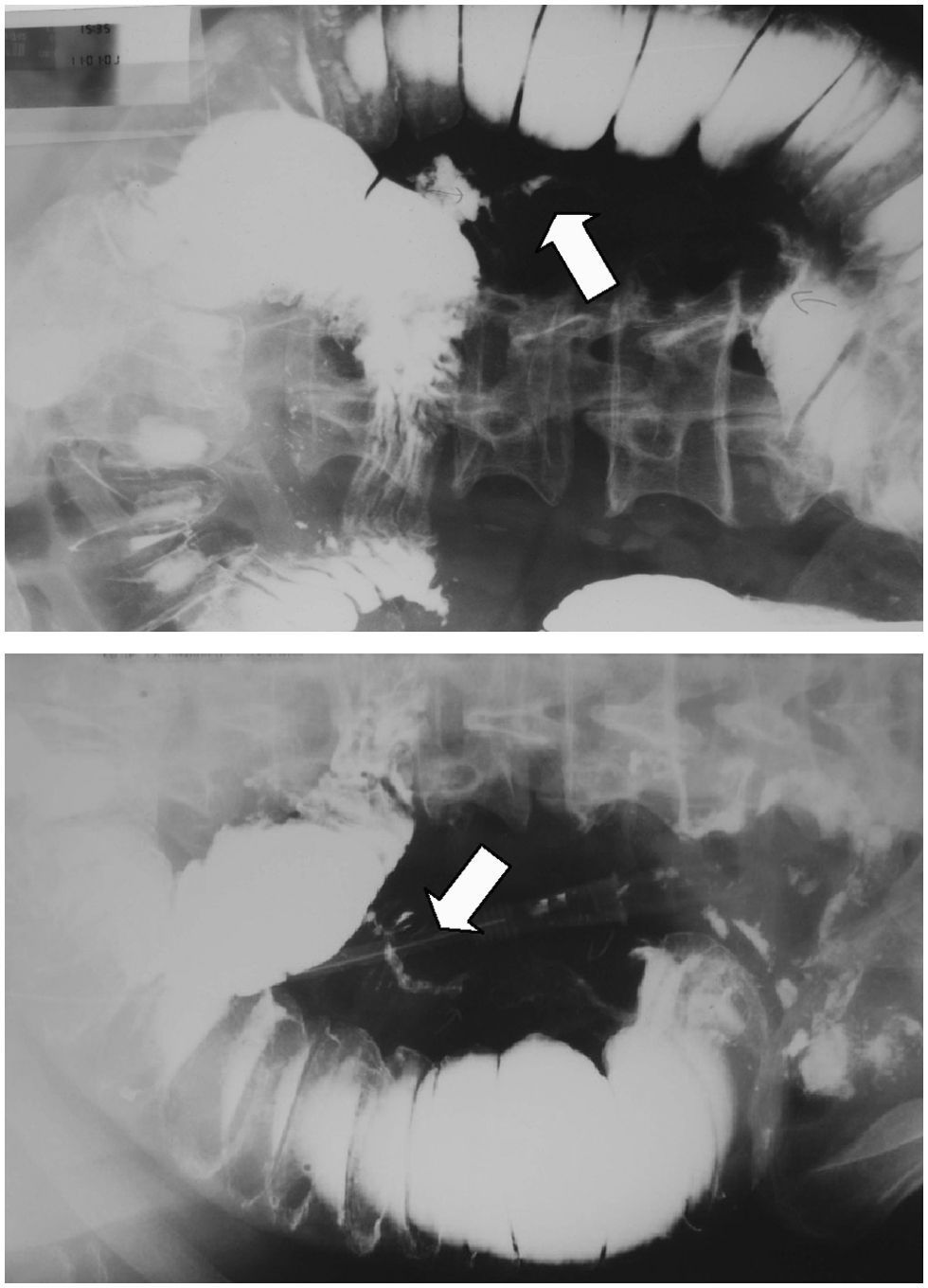
En el examen radiológico del abdomen, se apreciaban asas del intestino delgado dilatadas (fig. 1), al igual que en la ecografía abdominal, en la que las asas fueron descritas como hipoactivas y con contenido en su interior. Se practicó un transito barritado que mostró una estenosis en el segmento medio del intestino delgado, con paso filiforme del contraste (flechas de la fig. 2).


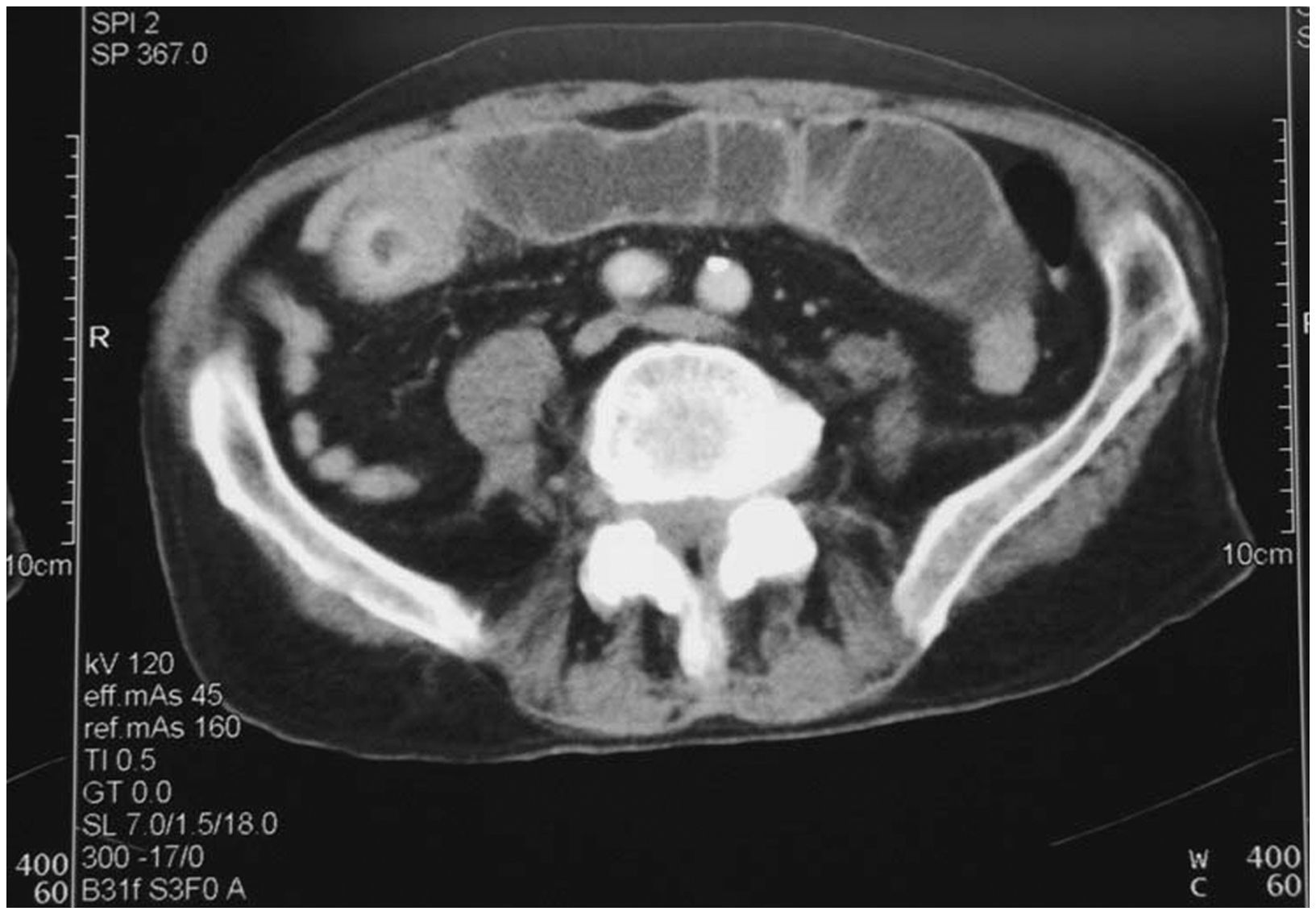
En una tomografía computarizada urgente se confirmó la dilatación de asas del yeyuno con engrosamiento de la pared intestinal y efecto de masa intraluminal (fig. 3). Al ser valorados por cirugía, se practicó laparotomía y se comprueba la existencia de un tumor en el segmento medio del yeyuno de unos 7cm, sin adenopatías ni lesiones tumorales hepáticas, y se practica una resección con anastomosis término-terminal.

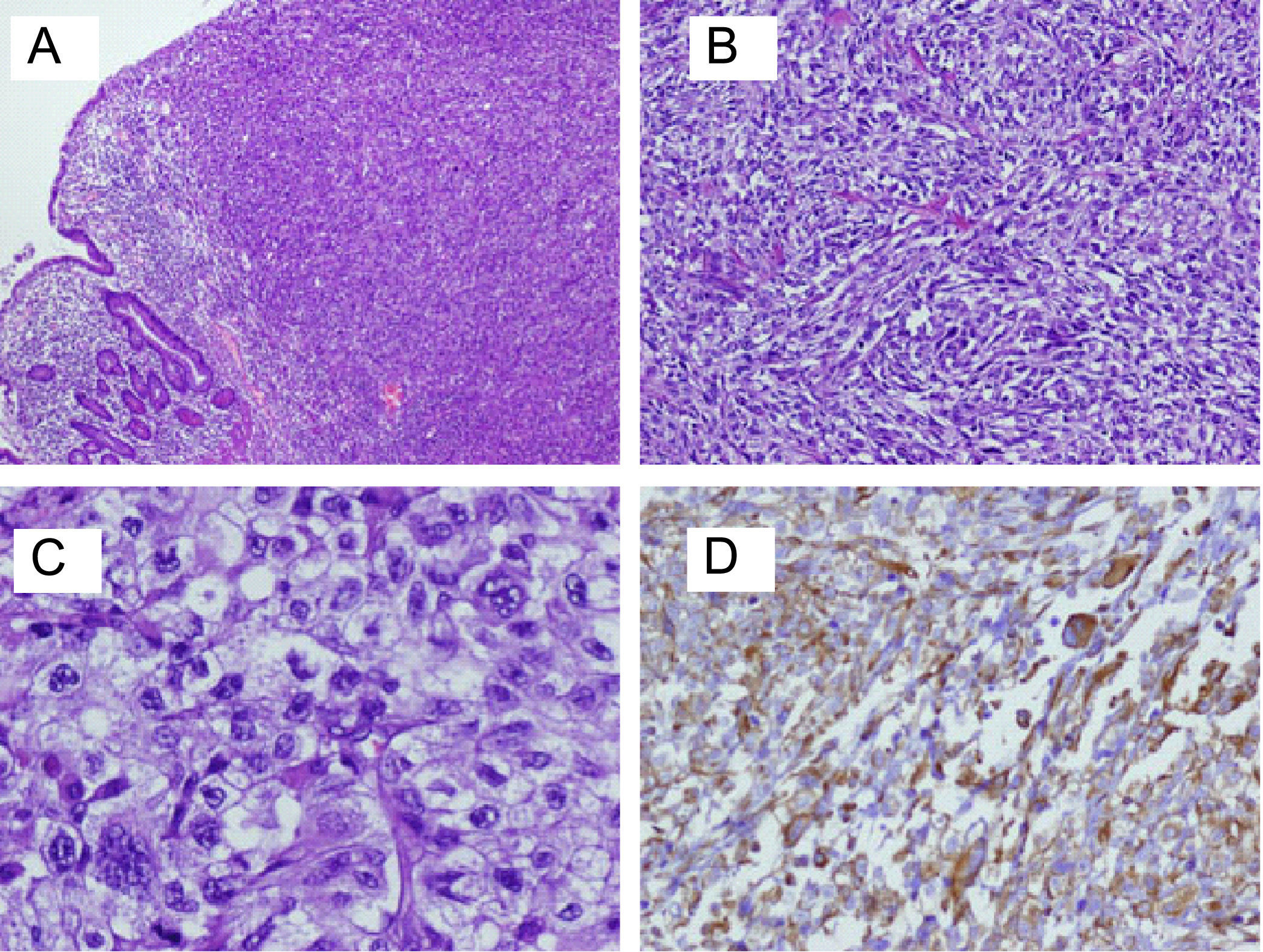
Para el estudio anatomopatológico, se recibió una pieza de 230mm de longitud, que presentaba una tumoración circunferencial de 50×30mm, parcialmente ulcerada, que infiltraba 10mm la pared, dando lugar a estenosis marcada de la luz. En el examen microscópico, se observó una tumoración constituida por una proliferación bifásica de células fusiformes y células epiteliodes, con marcada atipia, numerosas mitosis y áreas de necrosis, que se disponían formando haces que se entrecruzaban (fig. 4A–C). La tumoración invadía toda la pared muscular del intestino llegando a la grasa subserosa. Los márgenes quirúrgicos estaban libres de tumor. Se aislaron 18 ganglios del mesenterio sin que se observara metástasis en ninguno de ellos. El diagnóstico emitido fue de carcinoma sarcomatoide pobremente diferenciado, con invasión de subserosa.

En el estudio inmunohistoquímico, se observó positividad para marcadores propios de los tumores epiteliales, como la citoqueratina AE1-AE3 o antígeno epitelial de membrana (EMA), así como positividad en las mismas células, para marcadores propios de las células sarcomatosas, como la vimentina (fig. 4D). A su vez, las células tumorales mostraban pérdida de marcadores propios de las células intestinales, como la citoqueratina 20, debido a un proceso de indiferenciación. Otros marcadores, como actina músculo liso, desmina, CD117, CD34, fueron negativos, lo cual descartaba otros sarcomas, como tumores del estroma gastrointestinal y leiomiosarcoma.
El curso postoperatorio fue desfavorable al presentar el paciente un cuadro séptico con fallo multiorgánico y refractariedad a las medidas terapéuticas habituales (fluidoterapia, aminas y drotrecogina alfa).
DiscusiónEl término de carcinoma sarcomatoide se utiliza en la reciente clasificación de la Organización Mundial de la Salud para neoplasias malignas que pueden tener diferenciación bifásica, epitelial y mesenquimal (con o sin componentes heterólogos). Las localizaciones más habituales suelen ser la cabeza, el cuello, el aparato respiratorio, las glándulas mamarias, las glándulas salivares y tiroides, el sistema urinario (el riñón y la vejiga) y la piel. La localización en el ámbito gastrointestinal es infrecuente5,6. De estas últimas, la localización en orofaringe y de esófago son las más habituales (aunque suponen menos del 5% de las neoplasias esofágicas)7 seguida por la localización en estómago (aproximadamente 37 casos publicados hasta el momento). Son excepcionales las localizaciones en el ámbito de hígado, páncreas, vía biliar, intestinos delgado y grueso. Hasta la fecha se han publicado solamente 20 casos de carcinoma sarcomatoide de localización semejante en el intestino delgado. La ubicación de los tumores ha sido en íleon (11 casos), yeyuno (8 casos) y un único caso de localización en el ámbito del duodeno5.
Los carcinomas sarcomatoides fueron descritas por primera vez por Wirchow en 18646,8. Posteriormente, en el año 1973, en la revista Gastroenterology, Dikman y Toser describieron por primera vez un carcinoma sarcomatoide de intestino delgado bajo la denominación inicial de enteroblastoma5,9. Desde entonces se han utilizado más denominaciones para la misma entidad tumoral, como por ejemplo carcinoma metaplásico, carcinoma de células fusiformes, seudosarcoma, carcinoma sarcomatoide o carcinosarcoma.
Son tumores que suelen aparecer en pacientes con una media de edad de 57 años. Al revisar la bibliografía, aparentemente el caso del paciente más joven publicado es de un varón de 35 años5. La distribución por sexo es de varón:mujer 1,5:1. No se ha relacionado con factores de riesgo o predisposición, aunque de los 20 casos publicados, en 2 ocasiones hubo antecedentes de enteritis de evolución larga5,10.
La presentación clínica habitual suele estar determinada por las complicaciones locales: dolor abdominal sordo, a veces de tipo cólico, obstrucción intestinal o hemorragia digestiva. Un porcentaje importante de pacientes presenta anorexia y pérdida de peso. También pueden iniciarse con manifestaciones metastásicas (está descrito un caso de síndrome de vena cava superior por metástasis linfática en el ámbito torácico)3. En todo caso, la identificación del tumor suele ser tardía, ante la expresividad clínica inespecífica del intestino delgado, lo que contribuye sin duda a una peor evolución posterior. El diagnóstico diferencial clínico es muy amplio al abarcar toda la enfermedad del yeyuno-íleon, siendo las pruebas de imagen y/o endoscópicas el procedimiento diagnóstico habitual, como en el caso presentado, aunque no es raro que se precise la intervención mediante laparotomía para llegar al diagnóstico final.
Desde el punto de vista microscópico, el carcinoma sarcomatoide se caracteriza por la coexistencia de 2 componentes celulares malignos: uno predominante de células epiteliales carcinomatosas, cúbicas o columnares de núcleo hipercromático, y otro de células mesenquimales fusiformes, pleomórficas, de disposición sarcomatosa (con núcleo ovalado, nucléolo prominente, citoplasma eosinofílico, y frecuente actividad mitótica), que además, se puede acompañar de un componente con diferenciación rabdomioblástica, osteoblástica, cartilaginosa, grasa o neuroblástica.
En el estudio inmunohistoquímico puede apreciarse positividad para diversos marcadores, entre ellos citoqueratinas, antígeno carcinoembrionario (CEA), EMA, propios de la célula epitelial, y vimentina, propio de la célula mesenquimal7,11.
Se han propuesto varias teorías acerca de las características histológicas del carcinoma sarcomatoide. En primer lugar, está la hipótesis sobre un tumor de «colisión», que indica la existencia de una proliferación simultánea, pero separada, de células epiteliales malignas y mesenquimales como respuesta a un estímulo desconocido. En segundo lugar, está la hipótesis sobre la inducción de estroma/metaplasia/fusión que sugiere que estos carcinomas aparecen al inducirse una transformación sarcomatosa en el estroma adyacente12. Por último, existe la teoría de que las células stem multipotentes son capaces de diferenciarse tanto en tejido epitelial, como en tejido mesenquimal, siendo esta última hipótesis la más aceptada en la actualidad. Esta interpretación «clonal» se sustentaría en los análisis ultraestructurales de microscopia electrónica, los estudios con cultivos celulares in vitro y las características inmunohistoquímicas con marcadores específicos de diferenciación epitelial y/o mesenquimal5.
En el diagnóstico diferencial de los tumores malignos de intestino delgado es imprescindible incluir los tumores benignos, los adenocarcinomas, los sarcomas, los tumores metastásicos, el histiocitoma fibroso maligno y los tumores estromales gastrointestinales (leiomiomas, leiomioblastomas y leiomiosarcomas).
Los carcinomas de intestino delgado, en general, presentan un pronóstico reservado (37–63% supervivencia a los 5 años) que empeora en el caso del carcinoma sarcomatoide, por lo avanzado en el momento del diagnóstico. De los 20 casos previamente publicados, un 70% de los pacientes falleció entre 2 meses hasta 3 años después del diagnóstico. De ellos, un 79% presentaba en el momento de fallecer un tumor recurrente o extensión metastásica de tipo carcinomatoso, sarcomatoso o mixto5, y es común que las metástasis linfáticas sean predominantemente de tipo carcinomatoso.
El pilar principal del tratamiento es la resección quirúrgica, ya que los pacientes presentan mínima respuesta, casi nula, a quimioterapia (5-FU, dexorrubicina, cisplatino, etc.) y/o radioterapia13,14.
La evolución fulminante es muy característica de estos tumores, con rápida metastatización. El tratamiento debe ser instaurado lo más temprano posible, y el pronóstico, como en nuestro caso, es muy reservado.
Cabe preguntarse si pudiera haber alguna relación casual entre la artritis reumatoide con síndrome de Sjögren presentado por el paciente y el tumor. Se sabe que la enfermedad inmunológica predispone a la aparición de neoplasias, pero los más comunes son los linfomas, generalmente después de varios años de evolución. Por el contrario, son más raros los tumores epiteliales y, tras una revisión en Medline (Pubmed) utilizando las palabras «Sarcomatoid tumor» y «Sjögren disease», hasta el presente no hemos encontrado ningún caso previamente publicado. De igual forma, cabe especular si la enfermedad inmunológica de base contribuyó al desenlace desfavorable con sepsis refractaria.

