




La enfermedad de Wilson (EW) es una enfermedad congénita de herencia autosómica recesiva, que se caracteriza por una reducción de la excreción biliar de cobre y se manifiesta por una acumulación de este metal, primero en el hígado y más tarde en el cerebro, donde ejerce un efecto tóxico1. La enfermedad se debe a una disfunción de la proteína que interviene en el transporte intrahepatocitario del cobre, hecho que impide que el cobre sea excretado al canalículo biliar. En un 90% de los casos la alteración de esta proteína también impide la incorporación del cobre a la aceruloplasmina, por lo que su concentración está reducida en el suero2.
Clínicamente se manifiesta como una enfermedad hepática, una enfermedad neurológica o neuropsiquiátrica, o como una combinación de ambas. El diagnóstico precoz es fundamental, dado que la lesión que produce en los diferentes órganos es acumulativa. En la actualidad se dispone de alternativas terapéuticas eficaces que impiden la progresión de la enfermedad si el diagnóstico se efectúa en fase temprana3.
La enfermedad incide fundamentalmente en niños, adolescentes y adultos jóvenes, por lo que no suele pensarse en ella en los pacientes de mayor edad. Sin embargo, algunas publicaciones recientes señalan que un significativo número de pacientes se diagnostican después de los 40 años, algunos porque sus síntomas se presentaron después de esta edad y otros porque no habían sido adecuadamente diagnosticados con anterioridad4–6.
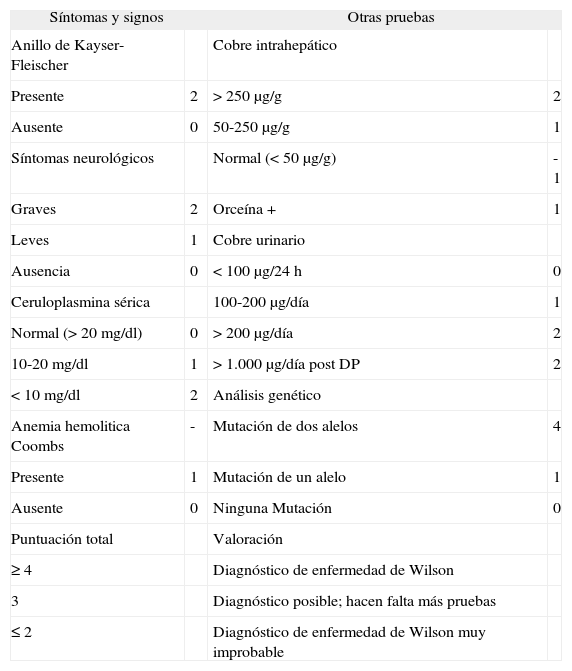
El diagnóstico clínico de la EW no es difícil cuando los pacientes muestran una expresión completa, como un descenso de la cifra de ceruloplasmina, una excreción elevada de cobre en la orina, la presencia de anillo de Kayser-Fleischer en el examen ocular, pero más del 10% de los pacientes presenta una ceruloplasmina en suero y una cupruria normales, y más del 50% con enfermedad hepática sin clínica neurológica no presenta anillo de Kayser- Fleischer7–9. En estos casos, si se mantiene la sospecha de enfermedad de Wilson, debe procederse a la cuantificación del cobre intrahepático. Unos valores superiores a 250 μg/g de tejido hepático seco son un indicador diagnóstico de EW si se ha excluido una colestasis crónica, que también cursa con una concentración intrahepática de cobre elevada, ya que en estos casos se produce un bloqueo de la excreción biliar del cobre, que es el mecanismo fisiológico para eliminar el exceso de cobre no utilizado por el hígado10. Si no se puede efectuar la medición del cobre intrahepático, el diagnóstico en los casos difíciles exige la prueba genética de una mutación del gen ATP7B. El análisis genético está muy limitado en la práctica porque la mayoría de pacientes con EW son heterocigotos compuestos, y se han identificado más de 200 mutaciones distintas de este gen11. Para superar las dificultades diagnósticas de la EW, un grupo de expertos ha propuesto utilizar un sistema de puntuación en el que se dan valores distintos a determinadas características de esta enfermedad (tabla I)12.
Sistema de puntuación propuesto para el diagnóstico de la enfermedad de Wilson
| Síntomas y signos | Otras pruebas | ||
| Anillo de Kayser-Fleischer | Cobre intrahepático | ||
| Presente | 2 | > 250 μg/g | 2 |
| Ausente | 0 | 50-250 μg/g | 1 |
| Síntomas neurológicos | Normal (< 50 μg/g) | -1 | |
| Graves | 2 | Orceína + | 1 |
| Leves | 1 | Cobre urinario | |
| Ausencia | 0 | < 100 μg/24 h | 0 |
| Ceruloplasmina sérica | 100-200 μg/día | 1 | |
| Normal (> 20 mg/dl) | 0 | > 200 μg/día | 2 |
| 10-20 mg/dl | 1 | > 1.000 μg/día post DP | 2 |
| < 10 mg/dl | 2 | Análisis genético | |
| Anemia hemolitica Coombs | - | Mutación de dos alelos | 4 |
| Presente | 1 | Mutación de un alelo | 1 |
| Ausente | 0 | Ninguna Mutación | 0 |
| Puntuación total | Valoración | ||
| ≥ 4 | Diagnóstico de enfermedad de Wilson | ||
| 3 | Diagnóstico posible; hacen falta más pruebas | ||
| ≤ 2 | Diagnóstico de enfermedad de Wilson muy improbable | ||
Tomado de Ferenci et al12.
La importancia de hacer el diagnóstico correcto de la EW es evidente, ya que si dejamos sin tratamiento a un paciente con esta enfermedad se expone a un fallecimiento prematuro como consecuencia de alguna complicación de la enfermedad, pero si hemos diagnosticado de EW a un paciente que no la tiene, le forzamos a hacer un tratamiento innecesario durante toda su vida.
El diagnóstico de EW facilita el examen de los familiares de primer grado del paciente, entre los cuales podemos diagnosticar en fase presintomática la enfermedad y aplicar un tratamiento que impida la aparición de lesiones en el hígado o en el cerebro13.
No se conocen las causas de la gran variación fenotípica de la EW. Los estudios de correlación entre diversas mutaciones del gen ATP7B y la forma de manifestarse la enfermedad no han dado resultados convincentes14, por lo que se considera que factores ambientales u otros factores genéticos puedan desempeñar algún papel. Probablemente, un factor que ha limitado un mejor conocimiento de la EW es el limitado número de casos que la mayoría de especialistas en enfermedades del hígado tendrá la oportunidad de atender a lo largo de su vida profesional, y también al limitado número de casos que puede reunir un servicio especializado15–20. Estas limitaciones sólo pueden ser superadas por estudios multicéntricos internacionales.
El proyecto Euro-Wilson se ha diseñado recientemente con la finalidad de crear un sistema que permita obtener la máxima información posible sobre la EW a partir de una base de datos que incluya a todos los pacientes diagnosticados en los países de la Unión Europea (UE). El proyecto exige la uniformidad del sistema de recogida de datos, lo que indirectamente puede ejercer una influencia positiva para una buena praxis clínica de los médicos involucrados en el estudio. Al mismo tiempo, puede facilitar el diseño de estudios terapéuticos y la obtención de información acerca de la incidencia y la prevalencia de la enfermedad, de los factores que influyan en la expresión fenotípica y su eventual correlación con el genotipo.
Los médicos que aporten sus casos a la base de datos general recibirán una información periódica, generada por el equipo coordinador, sobre la marcha del estudio. Los beneficios suplementarios para estos médicos serán la confirmación diagnóstica efectuada por un comité de expertos, que analizará la información facilitada.
En la práctica, los médicos que deseen participar en el proyecto Euro-Wilson, que deberían ser todos los que atienden a pacientes con EW, tendrán que ponerse en contacto con el coordinador español que le corresponda, la Dra. Mercé Vegara para pacientes adultos de cualquier región de España y para los niños de Cataluña, y para los pacientes pediátricos la Dra. Loreto Hierro para la región Centro, el Dr. Víctor Naas para Andalucía y las Dras. Carmen Rivas y Pilar Codorin para Valencia. Estos doctores asumieron esta función de coordinador regional en una reunión celebrada en enero de 2007 en el Hospital Infantil de La Paz, organizada por la Dra. Paloma Jara, en función de coordinadora nacional, a la que asistieron gran número de especialistas españoles interesados en esta enfermedad. Los coordinadores suministrarán la plantilla a cada médico que desee notificar un caso, que incluye la información clínica requerida y el código de cada paciente, procedimiento indispensable para garantizar la confidencialidad de los datos recogidos.
El proyecto está patrocinado por la UE, por lo que se dispone de fondos para diseñar un sofisticado sistema de seguridad informática y efectuar los estudios genéticos necesarios. Este editorial debe interpretarse como una llamada a los hepatólogos españoles para que incluyan en la EW todos los casos diagnosticados desde enero de 2005, y para ello deben ponerse en contacto con alguno de los coordinadores del estudio.

