




La enteropatía autoinmune (EA) es una causa muy poco frecuente de malabsorción que suele asociarse a presencia de autoanticuerpos circulantes, predisposición a fenómenos autoinmunes y se da con más frecuencia en la edad pediátrica.
El diagnóstico de esta entidad se basa en cuatro criterios, la presencia de diarrea crónica (más de 6 semanas), un cuadro clínico compatible con malabsorción, una histología específica, la exclusión de otras enfermedades que pueden cursar de forma similar y la presencia de autoanticuerpos específicos como pueden ser los anticuerpos entienterocito y anticélula caliciforme. Presentamos a continuación el caso de un paciente de 63 años de edad con un cuadro clínico que consiste en diarrea crónica, pérdida de peso e importante repercusión en su estado nutricional.
Autoimmune enteropathy (AIE) is an infrequent cause of malabsorption that is usually associated with the presence of circulating autoantibodies and a predisposition to autoimmune disorders. This disease is more frequent in children.
The diagnosis of this disorder is based on five criteria: chronic diarrhea (>6 weeks), malabsorption, specific histological findings, exclusion of similar disorders, and the presence of specific antibodies such as anti-enterocyte and anti-goblet cell antibodies. We present the case of a 63-year-old patient with chronic diarrhea, weight loss and significant deterioration of nutritional status.
Los cuadros de malabsorción se caracterizan por la diarrea crónica, la pérdida de peso y la malnutrición del paciente que los padece. Existen múltiples entidades que cursan con malabsorción, desde infecciones hasta enfermedades autoinmunes, pasando por tumores, enfermedad celíaca, enfermedad inflamatoria intestinal, etc.
En algunas ocasiones el diagnóstico es de exclusión y la histología de la biopsia de intestino delgado o grueso juega un papel muy importante.
Los criterios establecidos para el diagnóstico de la enteropatía autoinmune (EA) son:
- 1.
Diarrea crónica (más de 6 semanas).
- 2.
Malabsorción.
- 3.
Histología específica: atrofia vellositaria, linfocitosis en criptas profundas, aumento del número de cuerpos apoptóticos y mínimos linfocitos intraepiteliales.
- 4.
Exclusión de otras causas de atrofia.
- 5.
Presencia de anticuerpos específicos (anticuerpos antienterocito).
Los cuatro primeros criterios son necesarios, la ausencia de anticuerpos antienterocito no excluye el diagnóstico1–3.
Los esquemas de tratamiento no están bien establecidos debido a que no existen series de casos con gran número de pacientes, se basa en la inmunosupresión del paciente, bien sea con corticoesteroides o con inmunomoduladores o una combinación entre ambos1,3,4.
A continuación, exponemos el caso clínico de un paciente sin antecedentes de interés que presentó un cuadro de diarrea crónica con malabsorción, en el cual se excluyeron las causas más frecuentes de malabsorción y se llegó al diagnóstico de EA, una causa muy poco frecuente de diarrea crónica en la edad adulta.
Caso clínicoVarón de 63 años de edad, sin alergias medicamentosas conocidas, con hipertensión arterial y antecedentes de crisis renoureterales que acudió a la consulta externa de aparato digestivo por presentar cuadro de diarrea de más de 4 semanas de evolución, con 10–15 deposiciones líquidas sin productos patológicos al día, sin dolor abdominal, sin náuseas ni vómitos e importante pérdida de peso, unos 10 kilogramos desde el inicio del cuadro. En la exploración física destacaba una importante malnutrición del paciente. La auscultación cardiopulmonar y la palpación abdominal fueron normales y no se palpaban adenopatías en ninguna región.
Analíticamente destacaba una ligera elevación de la PCR (2,08mg/dl), una discreta disminución de la albúmina (2,9g/dl) con proteínas totales en rango normal (4g/dl), niveles disminuidos en el metabolismo lipídico (colesterol total 95mg/dl; triglicéridos 92mg/dl; HDL 22mg/dl) y una moderada ferropenia (hierro 29,3μg/dl; transferrina 133μg/dl; CFT hierro 169μg/dl e índice de saturación 17,3%). También presentaba una alteración del tiempo de protrombina (55%).
Se solicitaron cultivos seriados de heces para bacterias y parásitos, siendo negativos, y sospechando un cuadro de malabsorción, se solicitó D-xilosa, que resultó ser patológica (<15mg/dl), así como principios inmediatos en heces donde destacó una importante esteatorrea.
Las catecolaminas en orina, el ácido 5-hidroxindolacético (A5HIA), las inmunoglobulinas séricas, las porfirinas en orina, la gastrina basal, el Mantoux, las serologías para VIH, VHB, VHC, IgM para CMV y los marcadores tumorales (CEA, CA 19.9, PSA, beta 2 microglobulina) fueron todos negativos y la función tiroidea resultó del mismo modo normal.
Se realizaron los anticuerpos para la enfermedad celíaca mediante fluoro-enzimoinmunoanálisis (EIA-F) obteniéndose los siguientes resultados; Ac anti-gliadina IgA 2,8u/ml (0–7), Ac anti-gliadina IgG 1,2u/ml (0–7), Ac antiendomisio negativo (<15), Ac anti-transglutaminasa 1u/ml (0–7).
Las pruebas de imagen, ecografía abdominal y TC toraco-abdomino-pélvico no añadieron ningún dato relevante.
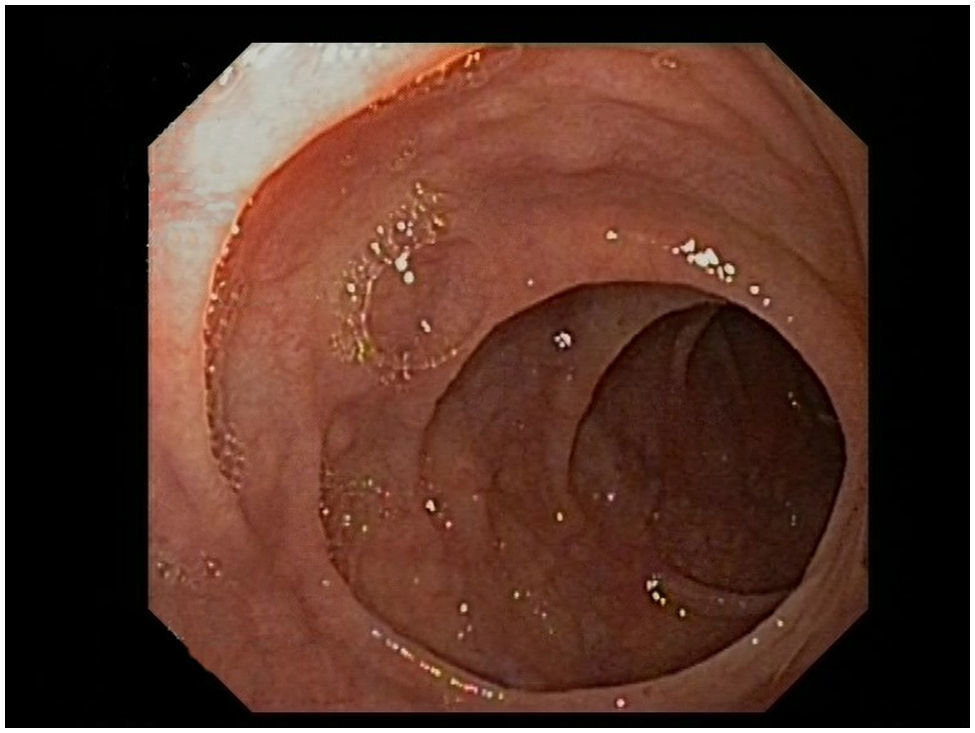
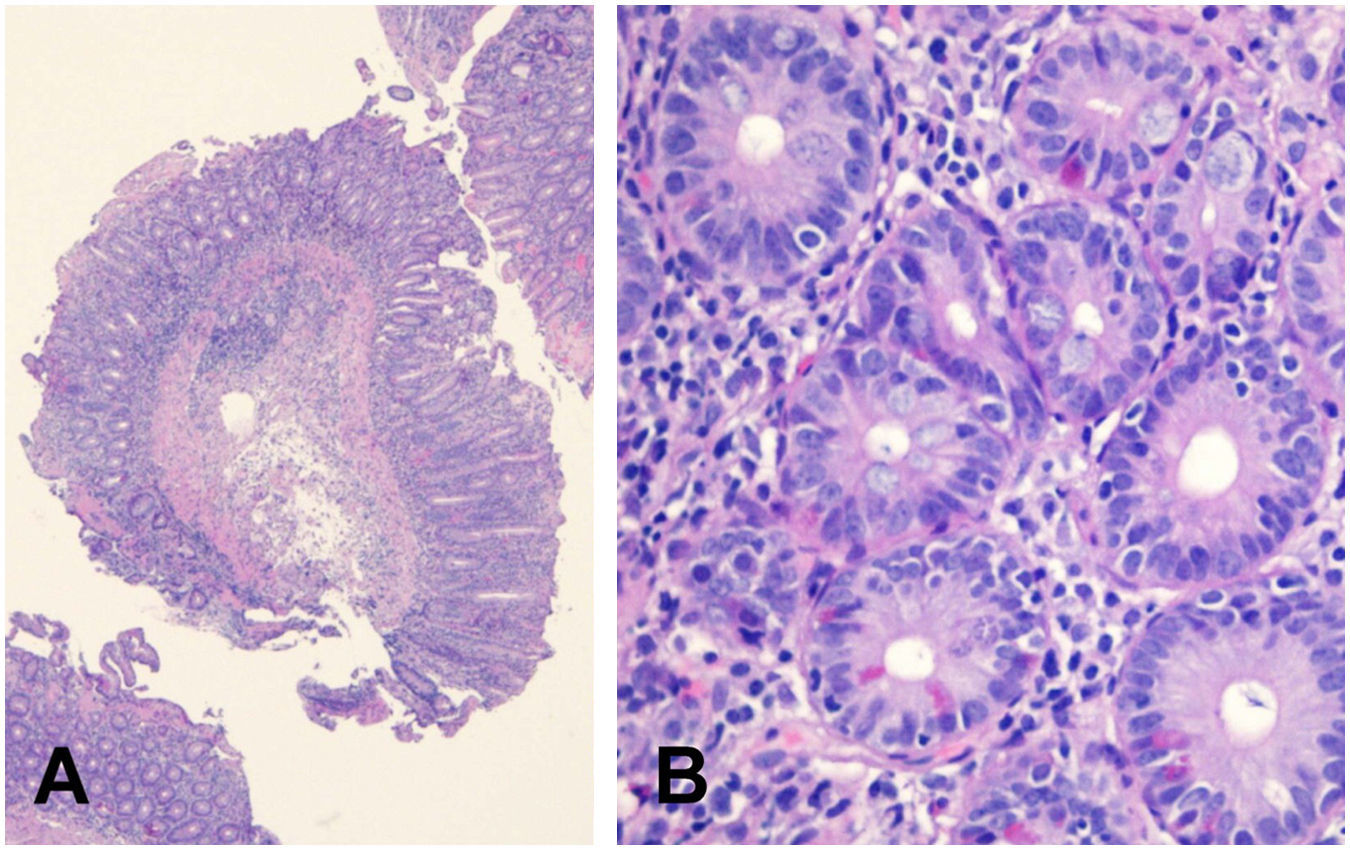
Se le realizaron gastroscopia y colonoscopia donde no se hallaron datos patológicos, por lo que se decidió realizar cápsula endoscópica donde se objetivó atrofia vellositaria de todos los tramos de intestino delgado. Posteriormente se hizo una enteroscopia parcial, en la cual se observó en un tramo de unos 50cm de yeyuno, una mucosa suavemente nodular, con placas blanquecinas en otras zonas, compatible con atrofia vellositaria (fig. 1), y se tomaron múltiples biopsias. Las biopsias tomadas del duodeno mostraban, de modo generalizado, una notable distorsión de la arquitectura, provocada por hiperplasia criptal acompañada de una atrofia vellositaria marcada, casi total en algunos fragmentos (fig. 2A). En el epitelio superficial no se apreciaba una linfocitosis significativa (12linfocitos/100 enterocitos), que sí se observaba en el epitelio criptal más profundo (>40 linfocitos/100 enterocitos) compuesta por linfocitos T, CD3+ y CD4+ (fig. 2B).

. B: Linfocitosis significativa a nivel del epitelio criptal profundo compuesta por linfocitos T, CD3+ y CD4+.")
La membrana basal estaba engrosada de modo difuso, y atrapaba con frecuencia capilares en su espesor. La lámina propia se encontraba expandida por un infiltrado linfoplasmocítico moderado, que focalmente se extendía a la submucosa, junto con un aumento del número de eosinófilos. El estudio inmunohistoquímico no mostró alteraciones significativas (linfocitos T CD3, CD4, CD8 positivos policlonales). No se realizó citometría de flujo de las biopsias intestinales, para estudio de subpoblaciones linfocitarias.
Se solicitó el tipaje HLA de clase ii mediante la técnica PCR-SSOr- Luminex, (HLADQ2, HLADR3, HLADR4, HLADQ8) que fueron todos negativos para ambos alelos.
A pesar de ello y sospechando una enfermedad celíaca seronegativa, se pautó una dieta sin gluten sin mejoría inicial pero que se mantuvo posteriormente durante 6 meses sin obtener clara mejoría. Durante su ingreso el paciente recibió nutrición mixta, parenteral y enteral, y continuó con el cuadro diarreico aunque mejoró su estado nutricional.
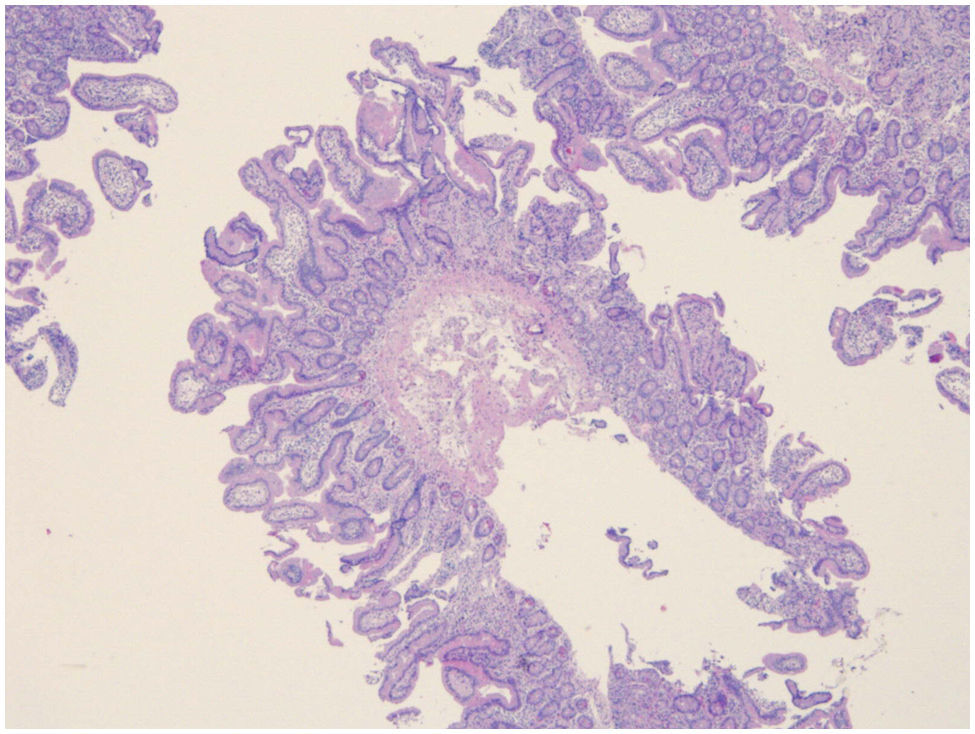
Con los resultados obtenidos, y con la exclusión de las principales causas de cuadros de malabsorción con atrofia vellositaria, sospechando una EA, se decidió comenzar tratamiento empírico con metilprednisolona a dosis de 1mg/kg/d, con gran mejoría del cuadro tras una semana de tratamiento con posterior retirada lenta y progresiva del mismo. En la actualidad, tras ocho meses de seguimiento, el paciente no recibe ningún tipo de tratamiento, ha ganado 10kg, realiza una deposición al día de características normales, ha experimentado una gran mejoría en su estado nutricional, presenta una D-xilosa normal e histológicamente, la arquitectura aparece parcialmente recuperada, con una atrofia vellositaria leve-moderada de distribución parcheada e hiperplasia criptal (9 linfocitos/100 enterocitos) (fig. 3) manteniendo un infiltrado linfocítico de la lámina propia, aunque más leve y focal, que ocasionalmente afecta al epitelio criptal profundo y al epitelio superficial, así como el aumento de eosinófilos y pequeños focos de actividad inflamatoria aguda.
Discusión
La EA es una causa muy poco frecuente de diarrea crónica, caracterizada además por pérdida de peso y malnutrición. Es más frecuente en la edad pediátrica y dada la rareza del trastorno, no existen datos suficientes sobre epidemiología, curso natural de la enfermedad y opciones terapéuticas. La serie de casos más larga publicada hasta el momento recoge 15 pacientes adultos con diagnóstico de EA. Según los datos obtenidos en esta serie, la edad media al diagnóstico fue de 55 años, el 47% eran varones y el 80% tenían asociados otros problemas de autoinmunidad como tiroiditis autoinmune, síndrome de Sjögren, artritis reumatoide, miastenia gravis, etc1. La predisposición a tener otras enfermedades autoinmunes se ha asociado con frecuencia a casos de EA. Sin embargo, la presencia de esta asociación no es realmente un rasgo que distinga esta entidad de otras patologías de intestino delgado, particularmente de la enfermedad celiaca4. Aproximadamente la mitad de los pacientes presentaban nódulos linfoides mesentéricos en el TC y el 53% presentaban alteraciones en la gastroscopia, incluyendo duodeno festoneado, mucosa en mosaico y atrofia vellositaria. La cápsula endoscópica presentaba atrofia vellositaria y mucosa en mosaico en el 71% de los casos1. Según los resultados de la Clínica Mayo, para realizar un diagnóstico de EA, son necesarios los siguientes criterios: la existencia de una diarrea crónica con malabsorción, una histología específica con atrofia vellositaria, linfocitosis en criptas, gran cantidad de cuerpos apoptóticos y mínimos linfocitos epiteliales, y la exclusión de otras causas de atrofia vellositaria5, fundamentalmente de la enfermedad celíaca, que es la causa más frecuente de aparición de atrofia de vellosidades. La presencia de autoanticuerpos específicos no es necesaria para realizar el diagnóstico ya que el significado en la fisiopatología de estos autoanticuerpos no está del todo clara6, su detección es observador dependiente y subjetiva y no existen análisis estandarizados para su detección. También se ha postulado en otros estudios que estos autoanticuerpos aparecen desde el inicio del daño de la mucosa y desaparecen después del tratamiento pero antes de que vuelva a ser una mucosa normal7,8.
La enfermedad celíaca (EC) y el daño intestinal de la inmunodeficiencia común variable se caracterizan por atrofia vellositaria e hiperplasia en las criptas, lo cual es muy parecido a lo que podemos encontrar en la EA9,10. La ausencia del haplotipo DQ2 o DQ8 del HLA puede ser útil para distinguir estos casos de una EC y sus formas refractarias, por su elevado valor predictivo negativo, ya que si son negativos, podríamos prácticamente excluir el diagnóstico de EC5,11. Aunque nuestro caso probablemente no se trate de una enfermedad celíaca refractaria Marsh 3b, hay que tener en cuenta que la respuesta a corticoides es totalmente inespecífica y se presenta en ambas enfermedades. La EC en el adulto es difícil de diagnosticar, pero sigue siendo la causa más frecuente con mucho de aparición de atrofia de vellosidades.
Por otro lado, la inmunodeficiencia común variable se caracteriza por la ausencia de células plasmáticas en las biopsias de intestino, y en la EA, puede aparecer un incremento de las células plasmáticas en la lámina propia. Histológicamente, la EA se distingue de la enfermedad celíaca por la carencia de un número significativo de linfocitos intraepiteliales, definido como >40 linfocitos epiteliales por cada 100 células epiteliales9.
La participación de gran variedad de autoanticuerpos intestinales y extraintestinales hace pensar que la EA representa un estado de hiperactividad del sistema inmune, que aparentemente ha perdido su propio autocontrol6. La fisiopatología de la EA no es muy bien conocida, pero la deficiencia o disfunción de las células T reguladoras CD25+ y CD4+ puede tener un papel importante. Las células T reguladoras CD35+ y CD4+ se encargan de mantener la autotolerancia inmunológica y disminuyen en varias enfermedades autoinmunes12. Estas células T reguladoras expresan específicamente el gen FOXP3, que codifica una proteína llamada escurfina12, cuya función es regular la supresión de la activación de las células T13. Las mutaciones de FOXP3 se han identificado en pacientes con enfermedades raras ligadas al cromosoma X que suelen presentarse en la edad pediátrica y abarcan desregulación autoinmune, poliendocrinopatía y enteropatía y se conoce como síndrome IPEX (de las siglas en inglés: Immunodysregulation Polyendocrinopathy Enteropathy X-linked síndrome)14. Existen gran cantidad de similitudes entre la EA y el síndrome IPEX. Aunque el síndrome IPEX es mortal en niños varones, se han observado variantes fenotípicas descritas en la edad adulta13. En cuanto al tratamiento, no existe un esquema terapéutico establecido. Según la experiencia de la Clínica Mayo, en todos los casos se utilizó tratamiento con corticoides asociado o no a azatioprina o infliximab, obteniendo una resolución completa de la diarrea en el 60% de los casos, una respuesta parcial en el 20% de los casos y ausencia de respuesta en el otro 20%1.
Se necesitan nuevos estudios e investigaciones para discernir si la EA es realmente una entidad clínica independiente o bien representa una variación de otra enfermedad del intestino delgado, lo cual llevará a la utilización de tratamiento específicos basados en la fisiopatología de la enfermedad.

