





La peritonitis bacteriana espontánea (PBE) es una complicación frecuente y grave de los pacientes con cirrosis y ascitis; con una mortalidad hospitalaria del 30-50% y un riesgo de recurrencia al año en los pacientes que sobreviven del 70%1. Su diagnóstico se basa en un recuento de leucocitos polimorfonucleares en líquido ascítico mayor de 250 células/µl; característicamente, es una infección monomicrobiana, causada en su mayoría por gérmenes gramnegativos de origen entérico, en especial Escherichia coli y Klebsiella spp. Se obtiene un cultivo del líquido ascítico positivo, que confirma el diagnóstico, en el 70% de los casos2.
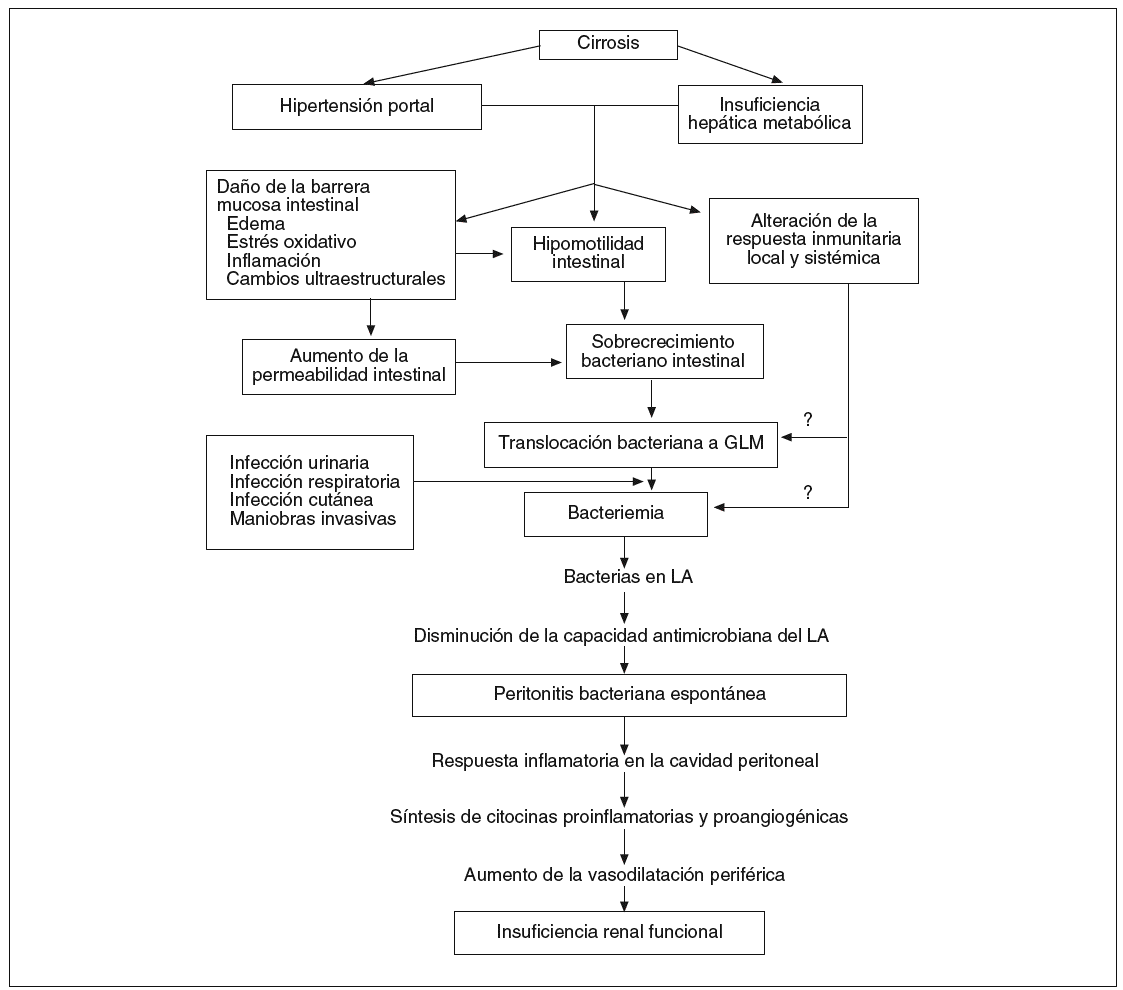
La hipótesis más aceptada en la actualidad para explicar la patogenia de la PBE es la colonización del líquido ascítico por gérmenes de origen intestinal, a partir de un episodio de bacteriemia3. Desde la luz intestinal, las bacterias cruzan la barrera epitelial mucosa e infectan los ganglios linfáticos mesentéricos (GLM), proceso conocido como translocación bacteriana (TB), y desde allí alcanzan la circulación sanguínea a través de la vía linfática. Los mecanismos por los cuales las bacterias intestinales alcanzan la circulación sanguínea y la linfa son aún desconocidos. Debido al intercambio constante de fluido entre la sangre y el líquido ascítico, las bacterias pueden pasar a la ascitis. Una vez que las bacterias han alcanzado el líquido ascítico, el desarrollo de la PBE depende en gran medida de la capacidad antimicrobiana del líquido. Los pacientes con una disminución de la capacidad defensiva del líquido ascítico desarrollarán peritonitis4.
Las observaciones clínicas iniciales que fundamentan esta hipótesis patogénica son: a) la mayor parte de las ocasiones, la PBE es una infección monomicrobiana, causada por bacilos gramnegativos de origen entérico2; b) el organismo causante de la infección puede ser aislado de cultivos de sangre periférica en el 50% de los pacientes con PBE con cultivos de líquido ascítico positivos4; c) la descontaminación intestinal selectiva con quinolonas disminuye la tasa de recurrencia de la PBE en un 71%5, y d) la probabilidad de desarrollar PBE en pacientes cirróticos con ascitis es mayor en los que presentan una disminución de la capacidad opsónica del líquido ascítico (identificada por una concentración total de proteínas < 1 g/dl en líquido ascítico) y/o una notable alteración de la actividad fagocítica del sistema reticuloendotelial (SRE) (identificada por la insuficiencia hepática avanzada)6. En un 10-20% de las ocasiones las bacterias proceden de otros focos diferentes del tubo digestivo, como la piel, la vía urinaria y la vía respiratoria. En los estudios clínicos más recientes se ha subrayado la importancia de la respuesta inmunitaria proinflamatoria del huésped, y su influencia en el deterioro del estado hemodinámico del paciente con cirrosis y ascitis, lo que constituye un factor decisivo en el pronóstico de la PBE7.
RELEVANCIA DE LA TRANSLOCACION BACTERIANA EN LA PATOGENIA DE LA PERITONITIS BACTERIANA ESPONTANEA EN LA CIRROSIS
El mecanismo patogénico clave que inicia la bacteriemia y posteriormente el desarrollo de la PBE es la TB. Ésta se define como la migración de microorganismos desde la luz intestinal a los GLM y a otros órganos extraintestinales8,9. Esta migración de bacterias es un fenómeno fisiológico normal, pero en individuos sanos el sistema inmunitario indemne es capaz de eliminar los escasos gérmenes que alcanzan los GLM. Para que la TB sea un fenómeno patológico, la migración de bacterias debe asociarse a una respuesta inflamatoria local o sistémica o con una posterior diseminación de las bacterias desde los GLM a la sangre o a la linfa. Así, la TB no sólo está aumentada en la cirrosis, sino también en situaciones asociadas a un alto riesgo de infecciones por bacilos gramnegativos y a fallo multiorgánico, como son el shock hemorrágico, la sepsis, la endotoxinemia, la obstrucción intestinal y los traumatismos graves10. En la cirrosis, el sistema inmunológico mucoso local puede ser incapaz de limitar no sólo el paso de bacterias a los GLM, sino también su proliferación en éstos. Las consecuencias de este hecho son dos: a) la diseminación de bacterias al torrente circulatorio, y b) el desarrollo de una reacción inflamatoria en los propios GLM con liberación de citocinas proinflamatorias.
Nuestro conocimiento sobre la patogenia de la TB deriva de estudios en modelos experimentales de enfermedad, dada la dificultad o imposibilidad de acceder a los GLM en los pacientes. Experimentalmente, la TB se define como un cultivo bacteriano positivo en los GLM11. La principal limitación de esta definición es que exige la presencia de bacterias viables en el GLM, pero muchas de las consecuencias de la TB pueden desencadenarse por productos bacterianos, como la endotoxina (lipopolisacárido [LPS]), cuya presencia no implica que haya bacterias viables. El diagnóstico de TB en clínica es complicado; por un lado, es difícil evaluar la translocación a los GLM debido a la ausencia de métodos no invasivos para su diagnóstico y, por otro, la mayor parte de los episodios de bacteriemia quedan sin diagnosticar. Recientemente, se ha propuesto la detección de ADN bacteriano en suero y líquido ascítico y de un aumento de la concentración sérica de proteína transportadora de LPS (LBP) como marcadores no invasivos de la TB11,12.
Los mecanismos que en condiciones normales evitan la TB son fundamentalmente tres: la estabilidad de la flora intestinal, la integridad del epitelio intestinal y las defensas inmunológicas del huésped. La TB representa una ruptura del equilibrio normal huésped-flora intestinal, que conduce a una respuesta inflamatoria continua, y finalmente a la infección.
El ecosistema bacteriano intestinal tiene una función importante en la prevención de la TB. Las vellosidades de la superficie apical epitelial están cubiertas de una capa mucosa, revestida de bacterias anaerobias. Estas bacterias previenen la adherencia a los enterocitos y limitan el sobrecrecimiento de bacilos aerobios gramnegativos. También los factores inmunológicos, como la secreción de IgA, previenen la adherencia de bacterias aerobias a los enterocitos. El sobrecrecimiento de la población entérica bacteriana gramnegativa o la reducción en la microflora anaerobia aumenta la susceptibilidad a la TB. Los factores endógenos que mantienen un equilibrio en la flora intestinal son la acidez gástrica, las secreciones pancrea tobiliares, los factores intestinales inmunológicos y, principalmente, la peristalsis intestinal. La barrera anatómica intestinal está compuesta de un epitelio de células columnares simples intercaladas con células especializadas, como células caliciformes, linfocitos y células presentadoras de antígeno. La conservación de la estructura y la función del epitelio normal, incluida la preservación de las uniones intercelulares, evitan la migración transepitelial o paracelular de bacterias. En situación normal, las bacterias contaminantes son fagocitadas por las células presentadoras de antígeno, y además hay otros mecanismos inmunológicos que contribuyen al aclaramiento bacteriano. La TB aparece en bajo grado en individuos normales. Un aumento marcado de los valores de TB y/o de la alteración de las defensas del huésped conlleva una replicación bacteriana en los GLM y su eventual diseminación a través de los vasos sanguíneos o linfáticos.
Un hecho destacable en la cirrosis es la coexistencia de la alteración en los 3 mecanismos de defensa13, lo que explica la alta tasa de TB observada en esta enfermedad. El daño simultáneo en los 3 pilares de la barrera intestinal propicia una mayor susceptibilidad a la TB respecto a otras situaciones clínicas o experimentales, en las que sólo está alterado uno de estos pilares14. Cada unos de estos mecanismos se ha estudiado en modelos animales y/o humanos de cirrosis.
Determinados estudios en modelos experimentales de cirrosis, generalmente inducida por tetracloruro de carbono, han señalado una tasa media aproximada de TB del 50% (rango, 40-80) en ratas con ascitis3,15,16. Este rango aumenta al 80-100% en los casos en que la ascitis está infectada8-9,15-17, y es del 30-60% en ratas con ascitis no infectada. De estas observaciones se puede deducir que la TB a los GLM es un paso inicial en el desarrollo de PBE17, lo que está en consonancia con los hallazgos clínicos en la cirrosis humana18,19. Por otra parte, el hecho de que un pequeño porcentaje de ratas con PBE no presente TB puede reflejar el origen no entérico de las bacterias, la baja sensibilidad de los métodos actuales para detectar microorganismos viables en los GLM y/o la capacidad inmunológica del huésped para aclarar las bacterias con translocación.
En la cirrosis, y particularmente en las formas más graves de enfermedad hepática y con historia previa de PBE, hay una mayor prevalencia de sobrecrecimiento bacteriano que en la población sana20. En ratas cirróticas se ha observado también que el sobrecrecimiento es más acusado en animales con TB que sin TB13. Este sobrecrecimiento se debe fundamentalmente a que en la cirrosis existe un tránsito intestinal enlentecido, favorecido por un aumento de la actividad adrenérgica y de la síntesis de óxido nítrico y por el daño oxidativo de la pared13,20,21.
La cirrosis está asociada a alteraciones funcionales y estructurales de la mucosa intestinal, que aumentan la permeabilidad a macromoléculas y bacterias. No está claro si los cambios estructurales, como la congestión mucosa, la inflamación submucosa y el edema son la causa o la consecuencia de la TB9. En casos de cirrosis experimental se ha demostrado que existe un estrés oxidativo de la mucosa del intestino delgado, con un aumento de la actividad de la xantin oxidasa y de la peroxidación lípidica del borde en cepillo de la mucosa intestinal22. Además, el defecto inmunológico en el aclaramiento de bacterias translocadas que se produce en la cirrosis está acompañado de una liberación de citocinas proinflamatorias inducida por endotoxinas12,23. La producción en cascada de factor de necrosis tisular (TNF-*) y óxido nítrico estimulada por endotoxinas agrava el daño oxidativo en el epitelio intestinal24. De los factores potencialmente implicados en la patogenia de la TB, la alteración en la flora intestinal es más determinante que la presencia de daño mucoso y la alteración de la permeabilidad intestinal13.
Para que la TB sea clínicamente significativa, aparezca PBE o bacteriemia, tiene que haber un defecto en las defensas locales y sistémicas del huésped. La relevancia de las alteraciones del sistema inmunitario intestinal en la patogenia de la TB y en la posterior diseminación de la infección está aún por definir. De hecho, la información disponible en la cirrosis se refiere a la función de las células inmunocompetentes circulantes, y este conocimiento se extrapola al sistema inmunitario intestinal. Diversos estudios in vivo e in vitro en modelos humanos de cirrosis avanzada han demostrado deficiencias en la capacidad bacteriostática y opsónica del suero, en la fagocítica de los neutrófilos, y en la función efectora de las células inmunocompetentes circulantes en sangre25-26. Además, la hiperemia esplácnica asociada a la hipertensión portal dificulta el rodamiento, la adherencia y la migración de las células fagocíticas en las vénulas mesentéricas27. Este conjunto de anomalías contribuye a facilitar la progresión y la diseminación de las infecciones locales en la cirrosis, provocando la bacteriemia. También es importante señalar que la actividad SRE se ve dañada en la cirrosis. El SRE es el arma más importante de defensa frente a bacteriemias adquiridas a través de la vía hematógena. En el hígado la mayor parte de la actividad del SRE asienta en las células de Kupffer. En los pacientes con cirrosis, la presencia de derivaciones portositémicas y de actividad fagocítica defectuosa de las células de Kupffer provoca una disminución en la actividad del SRE28.
La localización anatómica donde se produce la TB es objeto de discusión. Los primeros estudios sugirieron la ruta transcecal de la TB en la cirrosis9,15. Sin embargo, en algunos estudios más recientes13,21 se considera el intestino delgado como un lugar adicional de TB. En los modelos animales de cirrosis, más del 90% de las ratas con TB presentan sobrecrecimiento bacteriano en el yeyuno o el íleon, y la tasa de bacterias en ciego puede ser normal.
MODULACION DE LA TRANSLOCACION BACTERIANA EN LA CIRROSIS
Una gran parte de la evidencia que sustenta la relación causal entre TB e infecciones bacterianas proviene de estudios que demuestran que la disminución de la carga entérica de bacilos aerobios gramnegativos y del sobrecrecimiento bacteriano reduce la frecuencia de PBE. De acuerdo con la hipótesis actual de PBE, al disminuir la carga entérica de bacilos aerobios gramnegativos se debería reducir la tasa de TB, y con ello de PBE. Se ha demostrado que la administración de antibióticos orales poco absorbibles, principalmente quinolonas, disminuye la frecuencia de PBE en pacientes cirróticos de alto riesgo5. Sin embargo, en modelos murinos de cirrosis, el norfloxacino y el cotrimoxazol reducen la TB de organismos gramnegativos, pero no la tasa total de TB, debido al crecimiento secundario de cocos grampositivos29,30. Por otra parte, la aparición tan frecuente de bacterias fecales resistentes a quinolonas ha conducido a la búsqueda de alternativas diferentes a los antibióticos.
Estas alternativas incluyen la reducción de la carga de bacterias entéricas o de maniobras que atenúan el daño de la barrera mucosa intestinal13,21,31-36 (tabla I). Tanto cisapride como propranolol disminuyen la carga entérica bacteriana al disminuir el tiempo de tránsito intestinal13,21. En la cirrosis con ascitis hay una hiperactividad simpática que genera un retraso en el tiempo de tránsito intestinal a través de un adrenorreceptor ß-2; por ello, el bloqueo de esta ruta con propranolol puede acelerar la motilidad intestinal31. Los probióticos han dado lugar a resultados poco prometedores para reducir la tasa de TB32, cuando se han administrado para reequilibrar los valores de enterobacterias potencialmente patógenas y de anaerobios intestinales protectores. Lactobacillus ha sido capaz de colonizar adecuadamente el intestino de ratas cirróticas, pero también de translocarse a los GLM. Por otra parte, la administración a ratas cirróticas de ácidos biliares conjugados, que tienen capacidad bacteriostática, ha conseguido disminuir el sobrecrecimiento bacteriano, la endotoxinemia y las tasas de TB33. Por último, se ha indicado la capacidad de los antioxidantes, vitamina C y glutamato, para reducir la TB, la carga de bacterias entéricas y el estrés oxidativo de la pared intestinal de ratas con cirrosis34.

CONTRIBUCION DE LA TRANSLOCACION BACTERIANA EN LOS GANGLIOS LINFATICOS MESENTÉRICOS A LA ACTIVACION SISTÉMICA DEL SISTEMA INMUNITARIO-INFLAMATORIO EN LA CIRROSIS
El concepto actual de TB se fundamenta experimentalmente en el crecimiento de bacterias viables en el cultivo de GLM. Este concepto debería ampliarse para incluir productos bacterianos, como endotoxinas (LPS) o el ADN, capaces de interaccionar y estimular a las células del sistema inmunitario. Hay evidencias clínicas y experimentales recientes que indican que esta interacción entre células inmunitarias y productos bacterianos de procedencia entérica explicaría la activación del sistema inmunitario-inflamatorio que se produce en la cirrosis de forma sistémica, con un aumento en las concentraciones circulantes de citocinas proinflamatorias12,37,38.
La activación de la respuesta innata depende del reconocimiento específico de moléculas altamente conservadas en la superficie de los patógenos, que la mayor parte de las células mononucleares reconocen por medio de receptores de reconocimiento de patrón policlonales. En humanos, los receptores de proteína transmembrana tipo Toll (Toll like receptors [TLR]) regulan la inducción de la inmunidad innata frente a patógenos microbianos. En concreto, el TRL 4 es responsable de la producción de TNF-* en respuesta a endotoxinas, mientras que TLR 2 responde a productos derivados de gérmenes grampositivos. Se ha demostrado en pacientes con cirrosis una correlación significativa entre la expresión de TLR 2 en las células mononucleares y los valores de TNF-* y de receptores solubles de TNF-*39. Este hallazgo indica que, además del estímulo de bacterias gramnegativas y endotoxinas, también es importante el estímulo microbiano grampositivo en la producción de TNF-* y otras citocinas.
Se ha observado en pacientes y en modelos experimentales de cirrosis una profunda alteración en la distribución y el estado de activación de las células inmunitarias38,40. En los GLM de las ratas cirróticas se produce una expansión de monocitos y de la población de células T cooperadoras y supresoras38. Posteriormente, la recirculación de las células inmunitarias activadas en el GLM extiende la inflamación a la circulación sistémica. Sin embargo, en sangre periférica el compartimiento T, y en particular el de las células T vírgenes, se encuentra retraído, frente a un incremento en el porcentaje de células T activadas y una expansión de los monocitos circulantes38. El estado de activación de las células inmunitarias se caracteriza, tanto en pacientes como en modelos experimentales de cirrosis, por polarización Th1 de las células T a la producción de interferón-* y expansión y activación de los monocitos a la producción de TNF-*12,38.
La evidencia actual muestra que la activación del sistema inmunitario-inflamatorio en la cirrosis se debe en gran parte a la translocación de bacterias y productos bacterianos entéricos. Los estudios clínicos han constatado que este estado proinflamatorio sistémico es más acusado en los pacientes con paso frecuente de productos bacterianos a la circulación, e identificados por un aumento en la concentración sérica de LBP12 o por la presencia en sangre de ADN bacteriano41,42. Además, la descontaminación intestinal selectiva en pacientes cirróticos normaliza la concentración sérica de LBP y reduce las concentraciones circulantes de citocinas12. En ratas cirróticas, esta misma maniobra terapéutica atenúa la expansión de las células inmunitarias activadas a la producción de citocinas proinflamatorias (TNF-*, interferón-*) tanto en el GLM como en la sangre periférica38.
En conjunto, estos datos apoyan que en la cirrosis, la activación de las células inmunitarias circulantes se inicia en el mesenterio, cuando las células del sistema inmunitario allí residentes entran en contacto con las bacterias y los productos bacterianos de origen entérico. Una vez activadas, las células inmunitarias abandonan los GLM y por recirculación alcanzan la circulación sistémica extendiendo la inflamación.
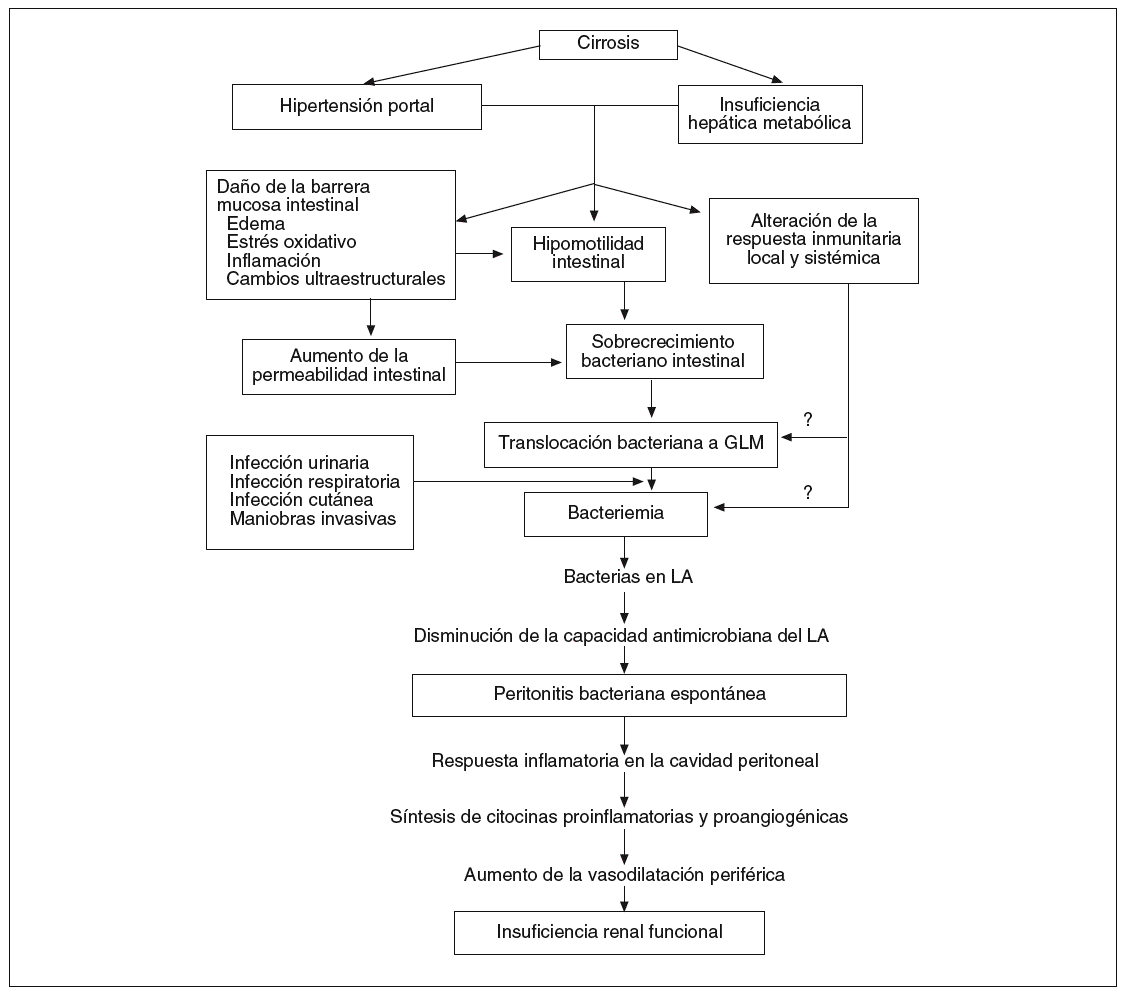
Fig. 1. Patogenia de la peritonitis bacteriana espontánea en la cirrosis. GLM: ganglios linfáticos mesentéricos; LA: líquido ascítico.
CONTRIBUCION DE LA TRANSLOCACION BACTERIANA Y DE LA ACTIVACION DEL SISTEMA INMUNITARIO-INFLAMATORIO SISTÉMICO A LA PROGRESION Y COMPLICACIONES DE LA CIRROSIS
La descripción pormenorizada de las consecuencias de la activación del sistema inmunitario-inflamatorio a la progresión, las manifestaciones clínicas y las complicaciones de la cirrosis escapan al alcance de esta revisión. El ejemplo mejor sustentado de esta relación entre el sistema inmunitario-inflamatorio y la complicación de la cirrosis lo constituyen las alteraciones hemodinámicas12,23. En gran medida, esta interacción se debe a la capacidad de las citocinas proinflamatorias, como el TNF-*, para estimular la sintetasa del óxido nítrico, y así contribuir a la regulación del tono vascular. De hecho, un aumento de TNF-* media el mayor deterioro hemodinámico que se observa en ratas y pacientes con cirrosis en los que se produce TB, expresado en las primeras por una mayor hiporrespuesta del lecho vascular mesentérico a vasoconstrictores y en los segundos por un grado mayor de vasodilatación periférica43,44. En pacientes con cirrosis, la descontaminación intestinal selectiva con norfloxacino atenúa la vasodilatación periférica y el estado circulatorio hiperdinámico43.
Las consecuencias de la TB también se extienden a la composición y a las características del líquido ascítico de los pacientes con cirrosis, que lejos de ser inerte, es biológicamente activo y con propiedades angiogénicas45. La activación de los macrófagos peritoneales por productos bacterianos, como LPS y ADN, y citocinas proinflamatorias, como interleucina (IL) 6 y TNF-*, estimula la síntesis de moléculas proangiogénicas, como el VEGF, y vasodilatadoras, como el óxido nítrico41,46-48. La sobreproducción de estas sustancias vasoactivas es mayor en los pacientes con ascitis infectada48, y puede contribuir a la vasodilatación arterial, la activación de los sistemas vasoactivos endógenos y la insuficiencia renal, que frecuentemente complica el curso de la PBE.
PATOGENIA DE LAS COMPLICACIONES DE LA PERITONITIS BACTERIANA ESPONTANEA
Aunque el pronóstico de la PBE ha mejorado en los últimos años, la tasa de mortalidad asociada persiste elevada, debido principalmente a la aparición de un fallo hepático avanzado y al desarrollo de complicaciones graves, como la insuficiencia renal, la hemorragia gastrointestinal y la encefalopatía hepática. La PBE es una causa común de insuficiencia renal en pacientes con cirrosis7. Un tercio de los pacientes con PBE desarrollará insuficiencia renal a pesar de la resolución de la infección7,49, y puede seguir 3 cursos clínicos diferentes. Un 15% de los pacientes con PBE desarrollará cambios en la función renal consistentes con un síndrome hepatorrenal tipo I. En estos pacientes, el deterioro rápido y progresivo de la función renal, a pesar de la resolución de la infección, se asocia a una mortalidad del 100%. En un 10-15% de los casos, la infección se asocia con un deterioro moderado y no progresivo de la función renal (síndrome hepatorrenal tipo II), que se estabiliza al resolverse la infección. En este caso la mortalidad hospitalaria es del 30-40%. Aproximadamente en un 10% de los pacientes con PBE la insuficiencia renal, es transitoria y revierte espontáneamente, y no puede ser considerada síndrome hepatorrenal. Este último curso clínico sólo presenta un 5% de mortalidad, similar a la de la PBE que cursa sin insuficiencia renal.
La causa del deterioro de la función renal podría deberse a una mayor disfunción circulatoria, causada por la propia infección y/o por los efectos deletéreos en la circulación renal de los productos bacterianos y las citocinas. La prevención del deterioro renal y la mejoría de la supervivencia en pacientes con PBE tras la expansión del volumen intravascular con albúmina intravenosa en el tratamiento de la PBE apoyan esta teoría50. El desarrollo de insuficiencia renal está asociado a valores plasmáticos muy elevados de TNF-* e IL6 y a un notable aumento de la actividad del sistema renina-aldosterona49,50. En los pacientes que desarrollan insuficiencia renal se observa que, durante el tratamiento y pese a la resolución de la infección, el gasto cardíaco y la presión arterial media disminuyen. En estas circunstancias, el gasto cardíaco no es capaz de compensar la mayor hipotensión arterial y reducción de la volemia efectiva, y se deteriora la perfusión tisular. Este deterioro afecta, entre otros, al riñón y al hígado, causando respectivamente, insuficiencia real y deterioro de la función hepática. Junto con ello, en estos pacientes se objetiva un marcado aumento de la presión portal50.
En resumen, la colonización del líquido ascítico por gérmenes de origen intestinal se inicia con la TB. Desde la luz intestinal las bacterias cruzan la barrera mucosa e infectan los GLM, y desde allí alcanzan la circulación sanguínea a través de la vía linfática. Los mecanismos por los cuales las bacterias intestinales alcanzan la circulación sanguínea y la linfa son aún desconocidos. Una vez que las bacterias alcanzan el líquido ascítico desde el torrente sanguíneo, el desarrollo de la PBE depende en gran medida de la capacidad antimicrobiana del líquido ascítico. Los pacientes con una disminución de la capacidad defensiva de éste desarrollarán peritonitis. La TB constituye el mecanismo patogénico clave inicial de la PBE. La coexistencia en la alteración de los 3 pilares de la pared intestinal al paso de bacterias, que se produce en la cirrosis, determina un aumento en las tasas de TB en estos pacientes. La TB, que es un proceso crónico, aunque fluctuante y episódico, somete al sistema inmunitario a una presión antigénica y a una exposición a productos con actividad inmunomoduladora de forma prolongada y recurrente, que conduce a una activación inadecuada de células accesorias, con producción alterada de mediadores solubles (citocinas y quimiocinas). En presencia de ascitis y de insuficiencia hepática grave, esta disfunción del sistema inmunológico y actividad proinflamatoria permite que las bacterias circulantes sedimenten en el líquido ascítico, produciéndose la PBE. Tanto las bacterias, como sus productos y las citocinas inducen la síntesis de óxido nítrico, lo que va a conducir a un aumento de la vasodilatación esplácnica y un agravamiento del estado circulatorio hiperdinámico. Cuando se infecta el líquido ascítico, y se desarrolla la PBE, la intensa producción de citocinas proinflamatorias en la cavidad peritoneal contribuye a agravar la vasodilatación periférica, favoreciendo la aparición de insuficiencia renal. La manipulación de la flora bacteriana intestinal y de su interacción con las células inmunitarias puede abrir nuevas expectativas en la profilaxis de la PBE y en la modificación del curso de la propia cirrosis hepática.

