




La neurofibromatosis tipo 1 (NF1) es una enfermedad multisistémica de herencia autosómica dominante, con penetrancia completa y expresividad variable. Afecta a uno de cada 3.000 individuos y se presenta en uno de cada 200 pacientes con retraso mental. Dentro de las múltiples complicaciones que asocia se encuentra la hemorragia digestiva secundaria a tumores mesenquimales gastrointestinales. Presentamos el caso de un paciente con NF1, ingresado por una hemorragia digestiva grave secundaria a un tumor yeyunal del estroma gastrointestinal (GIST), que presentó una buena evolución tras su exéresis quirúrgica.
Neurofibromatosis type 1 (NF1) is a multisystemic disease with an autosomal dominant pattern of inheritance, complete penetrance and variable expressivity. This disease affects one in every 3,000 individuals and one out of every 200 patients with mental retardation. Gastrointestinal bleeding secondary to gastrointestinal mesenchymal tumors is one of the many complications associated with NF1. We report the case of a patient with NF1 admitted to hospital for severe gastrointestinal hemorrhage secondary to a jejunal gastrointestinal stromal tumor (GIST) with favorable outcome after surgical removal.
Los pacientes con neurofibromatosis tipo 1 (NF1) presentan una elevada incidencia de neoformaciones a partir del tejido neuroectodérmico. En estos pacientes pueden aparecer tumores intestinales estromales, cuya situación y expresividad clínica dificultan a veces la realización de un diagnóstico y un tratamiento correctos. Aunque la mayor parte de la sintomatología de estos pacientes proviene del efecto mecánico compresivo de los neurofibromas sobre las raíces nerviosas, algunos de ellos pueden presentar síntomas digestivos específicos a modo de estreñimiento o derivados de la existencia de neoformaciones intestinales, sobre todo estromales. Estos tumores, según las recientes clasificaciones, se dividen en tres grupos: a) originados desde las células de Schwann; b) originados desde el músculo liso, y c) tumores yeyunales del estroma gastrointestinal (GIST). La aparición de GIST en el seno de la NF1 se ha observado en menos del 5% de los afectados por esta entidad, y la forma múltiple aún es más rara. Es importante tener presente la malignidad de los GIST (de hasta un 30%), así como las novedosas líneas de tratamiento molecular que, junto con la cirugía del tumor primario, consiguen excelentes resultados, aun en presencia de metástasis.
Observación clínicaVarón de 30 años de edad, sin alergias medicamentosas conocidas, con antecedentes de NF1, que requirió la colocación de una válvula de derivación ventrículo-peritoneal en la infancia a causa de una hidrocefalia, y cirugía para paliar una escoliosis en dos ocasiones. El paciente acude al servicio de urgencias por presentar deposiciones de tipo melénico asociadas a un cuadro sincopal, y refería una significativa pérdida de peso en los últimos meses. En la exploración física destacaba una presión arterial de 60/40mmHg y una frecuencia cardíaca de 113lat/min, la presencia de «manchas café con leche» en el tronco y las extremidades inferiores, así como una notable escoliosis dorsal. La auscultación cardiopulmonar y la palpación abdominal fueron normales. Analíticamente, destacaban los siguientes prámetros: hemoglobina 7,9g/dl; hematocrito 22%; volumen corpuscular medio 92,9fl; plaquetas 625.000/μl; leucocitos 12.400/μl, con un 70,2% de polimorfonucleares; razón normalizada internacional 1,08; tiempo de protombina 13 s; ALT 15U/l; AST 13U/l; bilirrubina 0,4mg/dl; GGT 22U/l; fosfatasa alcalina 86U/l; LDH 86U/l; creatinina 0,53mg/dl; urea 48mg/dl; proteínas 4,2g/dl; sodio 144mmol/l, y potasio 4,2mmol/l.
El paciente fue ingresado con el diagnóstico de hemorragia digestiva alta, con una gran repercusión hemodinámica y una analítica inicial con anemización progresiva (que requirió la transfusión de 4 concentrados de hematíes), por lo que se realizó inicialmente una panendoscopia oral urgente, sin que se apreciaran en ella restos hemáticos ni afectación a lo largo de todo el trayecto explorado. Ante estos hallazgos se decidió realizar una colonoscopia urgente, en la que se progresó hasta el polo cecal por el colon, con abundantes restos hemáticos, pero no se pudo apreciar la causa del sangrado.
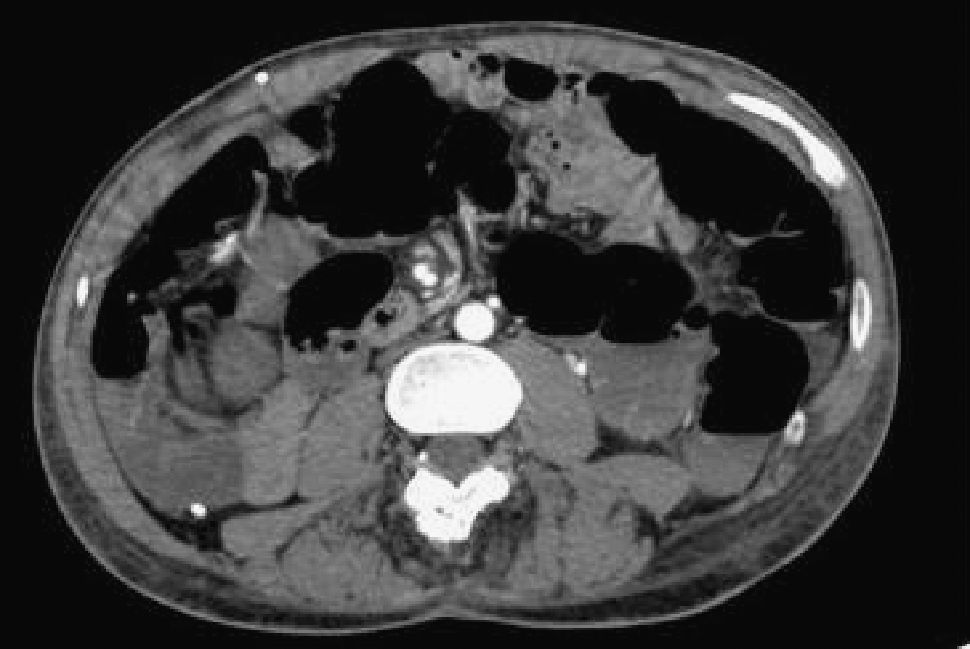
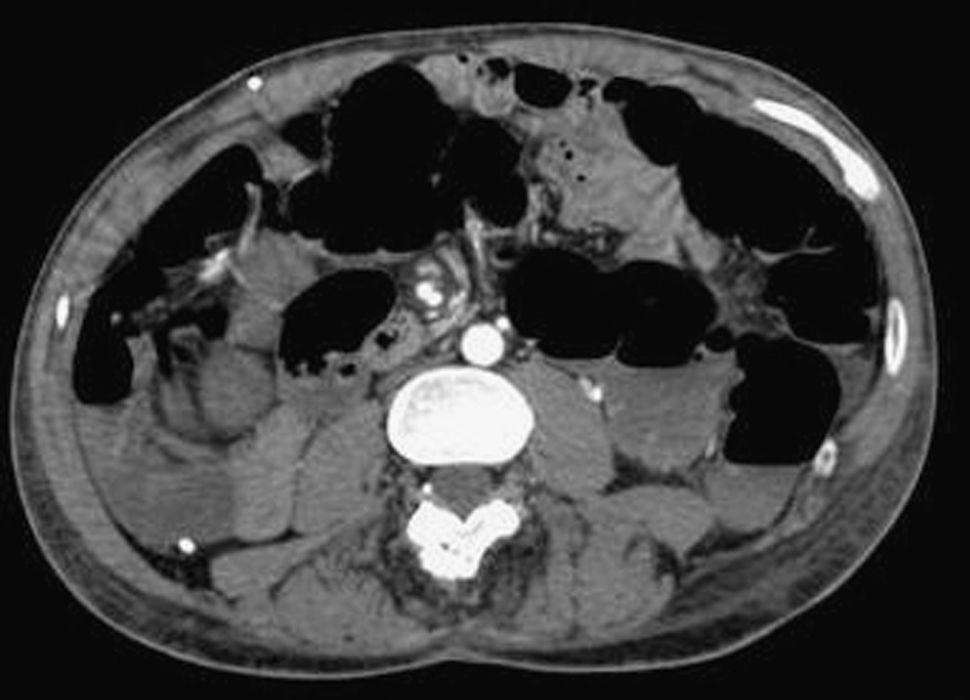
Dada la ausencia de punto sangrante en los procedimientos endoscópicos realizados inicialmente, la persistencia del sangrado, que comportaba la necesidad de continuar con las transfusiones sanguíneas, y los antecedentes del paciente, se realizó una tomografía computarizada (TC) abdominal urgente con los siguientes hallazgos (figs. 1 y 2): en el yeyuno proximal, inmediatamente posterior al ángulo de Treitz (aunque situado en el hipocondrio derecho por una anomalía en la rotación intestinal), se observaba una tumoración, con importante captación de contraste de aproximadamente 4,2cm de longitud y 4,5cm de espesor; dicha tumoración presentaba necrosis en su porción central, que era la causa de la hemorragia digestiva. Ante estos hallazgos, el paciente fue valorado por el equipo de cirugía de guardia, que decidió su intervención urgente. En la laparotomía se constató una tumoración en el yeyuno cercano a la cuarta porción duodenal, que engloba un segmento de íleon proximal, así como una dilatación colónica con contenido hemático en su interior. Se realizó una resección yeyunal de aproximadamente 10cm, que englobaba la tumoración, y una resección del íleon proximal de unos 10cm, con anastomosis enteroentérica lateral mecánica mediante EndoGIA 45. El paciente presentó una buena evolución posquirúrgica, con restablecimiento del tránsito y buena tolerancia a la reintroducción progresiva de la dieta oral. No se produjo ninguna exteriorización hemorrágica ni se observó anemización durante la hospitalización.
Histológicamente, se trataba de una tumoración de origen mesenquimal, compuesta por células fusiformes de citoplasma claro y núcleos ovalados de cromatina vesicular, con nucléolos evidentes. Se observaban áreas de celularidad escasa, con un fondo fibrótico, alternadas con áreas más celulares, con presencia de células de núcleos agrandados, de morfología muy irregular. Se aprecian aproximadamente tres figuras de mitosis por cada 50 campos de gran aumento. Los márgenes de resección quirúrgicos se hallaban respetados. El perfil inmunohistoquímico de las células tumorales muestra una positividad difusa para CD 117 (c-kit), CD 34 y vimentina, y negatividad para actina y S-100. El índice de proliferación celular es bajo (1%).
Para concluir, cabe afirmar que se trataba de un GIST de riesgo intermedio, que respetaba los márgenes quirúrgicos de resección.
DiscusiónLos GIST son las neoplasias mesenquimales submucosas más comunes del tubo digestivo. Representan el 0–1,3% de todos los tumores del tracto gastrointestinal, el 1–3% de las neoplasias gástricas, el 20% de las del intestino delgado y el 0,2–1% de las colorrectales1. Este grupo de tumores se originan en la muscular propia, entre ésta y la muscular de la mucosa, a partir de células madre primitivas, conocidas como células de Cajal. Histopatológicamente, se caracterizan por la expresión inmunohistoquímica del receptor transmembrana del protooncogén c-kit (y su proteína resultante CD 117), así como de la expresión del marcador CD34 (presente en el 70% de los tumores). Otros marcadores que pueden expresar los GIST incluyen la actina, la desmina y la proteina S-1002. Fisiopatológicamente, la mutación del protooncogén c-kit tiene un papel fundamental, ya que ésta permite la expresión de la proteína BCR-ABL, que es un receptor de superficie y transmembrana para un factor de crecimiento celular con función tirosina-cinasa. La oncoproteína BCR-ABL posee un compartimento para ATP que, por la acción de la tirosina-cinasa fosforiliza un sustrato que desencadena una cascada de fenómenos, los cuales terminan en una proliferación celular no controlada y resistencia a la apoptosis. La mutación de la proteína c-kit se presenta en el 52–85% de los casos3. Los principales factores de riesgo para su desarrollo son la NF1, presente en el 6,5% de los pacientes, la urticaria pigmentosa, y el raro síndrome familiar de GIST4.
La NF1 es una enfermedad progresiva, multisistémica, de herencia autosómica dominante, penetrancia completa y expresividad variable. La enfermedad afecta a uno de cada 3,000 individuos, y casi el 50% de los casos corresponden a neomutaciones. El gen de la NF1 se localiza en el cromosoma 17, en la banda q11 2. Dicho gen codifica la proteína neurofibromina, que se cree actúa como un supresor tumoral, regulando otra proteína celular que estimula el crecimiento y proliferación celular. Para el diagnóstico de NF1 el paciente debe poseer al menos 2 de los siguientes 7 criterios: 6 o más manchas «café con leche» iguales o mayores de 5mm en prepúberes y de 15mm en pospúberes; 2 o más neurofibromas de cualquier tipo, o uno plexiforme; presencia de pecas en las axilas o las ingles; glioma del nervio óptico; 2 o más nódulos de Lisch; lesión ósea definida como displasia del esfenoides o adelgazamiento de la cortical de los huesos largos con o sin seudoartrosis, y un familiar de primer grado afectado. Se pueden encontrar complicaciones derivadas de esta enfermedad en el sistema nervioso central (trastornos de aprendizaje, macrocefalia, convulsiones, déficit neurológicos secundarios a tumores, compresión medular), así como en el sistema nervioso periférico (neuropatía, tumores de nervios periféricos), cardiovascular (hipertensión arterial), endocrino (talla corta, trastorno neuroendocrino debido a tumores hipotalámicos, pubertad anormal, feocromocitoma), óseo (displasia de alas del esfenoides, escoliosis, arqueamiento congénito, quistes óseos, hipercrecimiento óseo), ocular (malformaciones orbitales, gliomas del nervio óptico) y gastrointestinal (neurofibromas intestinales).
En los pacientes con NF1, las manifestaciones clínicas de este tipo de tumores gastrointestinales son similares a las de los demás pacientes. Sin embargo, se presentan con mayor frecuencia en el intestino delgado y, generalmente, son múltiples, a diferencia del grupo sin factores de riesgo5. Cabe mencionar que pese a su conocida multiplicidad, el diagnóstico preoperatorio de la totalidad de los tumores es difícil mediante técnicas de imagen, dado su tamaño inferior a 1cm6. Hay cuatro formas de implicación digestiva: a) hiperplasia de la submucosa y de las células de Schwann de los plexos nerviosos mioentéricos, b) formación de diferentes variedades de tumores estromales; c) mayor prevalencia de carcinoides en la zona periampular, y d) mayor predisposición que la población general a padecer adenocarcinomas7. Las recientes técnicas inmunohistoquímicas han dividido con más certeza los tumores estromales digestivos a partir del tipo celular que los genera: en primer lugar, estarían los derivados de las células de Schwann, con componente de tejido neuronal, positividad para S 100 y negatividad para CD 117; es el caso de los neurofibromas, que pueden aparecer aislados o en el seno de la NF1. En segundo lugar, los formados a partir de células musculares lisas, con positividad para desmina-actina y negatividad para CD 117 y CD 34; en este grupo se incluirían los leiomiomas y leiomiosarcomas. Por último, como en el caso que nos ocupa, se hallarían los originados desde las células autónomas de Cajal, cuyo criterio diagnóstico principal es la positividad para CD 117 (c-kit) o CD 34, si bien se empiezan a describir algunos casos con estos marcadores negativos, como los GIST.
Desde el punto de vista clínico, las manifestaciones dependen de su tamaño y localización. Los tumores pueden cursar asintomáticos (incidentalomas) en el 50% de los casos. Sólo una tercera parte de las lesiones menores de 3cm cursan con síntomas, mientras que más de 50% de las lesiones mayores de 5cm tienen expresión clínica8. En general, en los pacientes sintomáticos, las formas de presentación son las siguientes: hemorragia del tubo digestivo secundaria a ulceración del tumor y síndrome anémico secundario, dispepsia, náuseas y vómitos, dolor abdominal, masa palpable, obstrucción intestinal con o sin intususcepción asociada y, rara vez, obstrucción renoureteral o como masa mediastínica con disfagia9. El tamaño varía entre milímetros y centímetros10. Se presentan como masas bien circunscritas sin cápsula verdadera y, dado que su origen es la capa muscular externa, tienen tendencia al crecimiento exofítico11, por lo que la apariencia más frecuente es la de un tumor que se origina de la pared intestinal y se proyecta hacia la cavidad abdominal. Pueden mostrar áreas de hemorragia, degeneración quística y necrosis. La ulceración del tumor se identifica en la mitad de los casos y la calcificación no es una característica habitual12. Las características observadas mediante técnicas de imagen son variadas y dependen del tipo de estudio radiológico. El 70-90% de los GIST son benignos, pero la posibilidad de malignidad difiere para cada órgano: los gástricos son casi siempre benignos, mientras que en el resto de los órganos el comportamiento suele ser más agresivo13. Los GIST malignos sólo representan el 3% de todos los cánceres del tracto gastrointestinal. En general, expresan la mutación de c-kit, son de mayor tamaño, y presentan mayor celularidad y mayor actividad mitótica que su contraparte benigna. Hay tres categorías de comportamiento histológico: probablemente benigno (menos de 5 mitosis por 50 campos de gran aumento y diámetro<2cm), indeterminado (menos de 5 mitosis por 50 campos de gran aumento y un diámetro de 3–4cm) y maligno (más de 5 mitosis por 50 campos de gran aumento y un diámetro igual o superior a 5cm)14. En un paciente con NF1 y clínica intestinal siempre se debe sospechar la posible existencia de neoformaciones digestivas. Sin embargo, el mayor reto es identificar la localización del sangrado digestivo. Es frecuente que la lesión no se encuentre en el trayecto explorado por gastroscopia y colonoscopia, por lo que en las hemorragias agudas se debe recurrir a técnicas isotópicas (cada vez menos usadas y con una sensibilidad de sólo un 52%), a la realización de endoscopia intraoperatoria (la sensibilidad puede aumentar al 70%) y, con mejor rentabilidad, el estudio de imagen a través de TC o a la arteriografía15. En las anemizaciones crónicas o los sangrados recurrentes, la realización de un tránsito intestinal o una enteroscopia podrían verse complementadas hoy día por el uso de la cápsula endoscópica16.
Actualmente, el tratamiento aceptado de los GIST es el quirúrgico. En caso de metástasis, los pacientes son candidatos a recibir mesilato de imatinib. Éste es un derivado de la fenilaminopirimidina, inhibidor selectivo de algunas proteincinasas: BCR-ABL, factor de crecimiento derivado de plaquetas y kit17. Este inhibidor ocupa el lugar destinado al ATP en el receptor BCR-ABL, necesario para la fosforilización dependiente de las tirosina-cinasas18,19.
El propósito de esta observación clínica es resaltar la importancia del diagnóstico diferencial y de la adecuada historia clínica de los pacientes para una correcta filiación etiológica de las entidades que padecen.

