





INTRODUCCIÓN
La hiperplasia de glándulas de Brunner (HGB) es una lesión duodenal disembrioplásica o hiperplásica infrecuente y una causa rara de hemorragia digestiva alta1-5. No se ha descrito hasta ahora la asociación de esta lesión con el síndrome antifosfolipídico. De hecho, en éste la afectación gastrointestinal es infrecuente6, la hemorragia digestiva es una complicación excepcional y la mayoría de los casos publicados son secundarios a lesiones isquémicas7,8. Presentamos un caso de hemorragia digestiva alta masiva por HGB en un paciente con síndrome antifosfolipídico primario (SAP).
OBSERVACIÓN CLÍNICA
Varón de 42 años de edad con antecedente de SAP diagnosticado a los 26 años a raíz de una trombosis venosa profunda de extremidades superiores, trombosis retiniana y anticoagulante lúpico y anticuerpos anticardiolipina IgG a títulos altos. En el momento del diagnóstico presentaba hipertensión arterial y deterioro de la función renal, con microangiopatía trombótica en la biopsia renal. A pesar de un correcto control de la presión arterial y del tratamiento anticoagulante, evolucionó a insuficiencia renal crónica terminal. Por este motivo requería hemodiálisis periódica desde hacía 11 meses y tenía anemia secundaria a la insuficiencia renal en tratamiento con eritropoyetina. Como otras complicaciones del SAP había presentado un episodio de trombopenia autoinmunitaria que se resolvió con tratamiento corticoide. Además tenía hiperuricemia con episodios de artritis gotosa y valvulopatía mitral con insuficiencia mitral de moderada a grave.
Ingresó en nuestro centro para recambio valvular mitral por válvula mecánica y durante la intervención requirió 4 concentrados de hematíes. En el postoperatorio se instauró inicialmente tratamiento anticoagulante con heparina de bajo peso molecular y posteriormente con anticoagulantes orales. Trece días después de la intervención presentó hemorragia digestiva en forma de hematemesis y melenas con shock hipovolémico. En el momento de ingresar en la unidad de hemorragias destacaban presión arterial de 80/47 mmHg, frecuencia cardíaca de 136 lat/min y temperatura de 37 °C. En la exploración física el paciente estaba consciente y orientado, con sudación fría e intensa palidez cutaneomucosa. Los tonos cardíacos eran regulares, sin soplos, con clic valvular audible y sin signos de fallo cardíaco. El abdomen era blando y sin dolor.
La analítica de urgencias mostró hematócrito del 22%, hemoglobina de 7,1 g/dl, velocidad corpuscular media de 90 fl, 22.300 x 109/l de leucocitos (el 90% neutrófilos, el 4% linfocitos y el 5% monocitos), 348.000 x 109/l de plaquetas, tiempo de protrombina del 6%, ratio normalizada internacional de 14,9, tiempo de tromboplastina parcial activada de 148/32, fibrinógeno de 4,3 g/l, glucosa de 125 mg/dl, urea de 356 mg/dl, creatinina de 12,5 mg/dl, Na de 129 mmol/l, K de 7,7 mmol/l, Ca de 8,7 mg/dl.
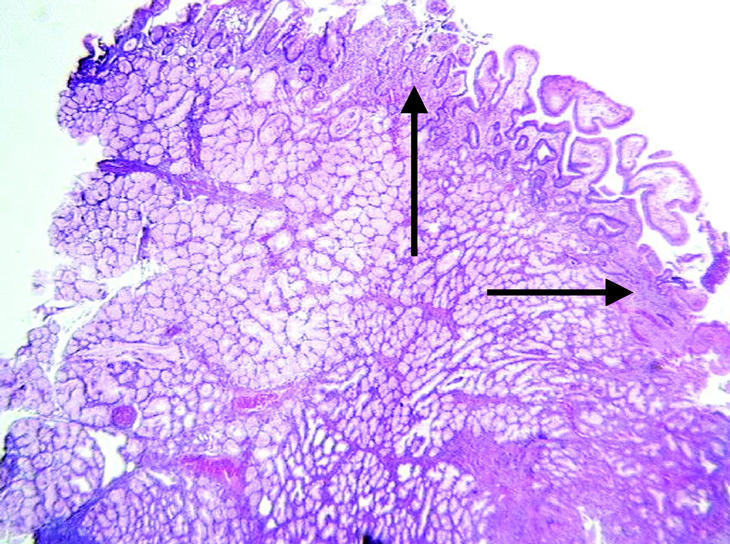
En el momento de su ingreso se inició transfusión sanguínea y de plasma fresco congelado (6 UI) y complejo protrombínico (2.400 UI; Prothromplex Inmuno Tim4), y se instauró tratamiento antisecretor con omeprazol por vía intravenosa. Presentó un episodio de fibrilación auricular que revirtió con amiodarona. Tras la corrección del exceso de descoagulación y transfusión de un total de 8 concentrados de hematíes se consiguió la estabilización hemodinámica y se realizó gastroscopia de urgencia, que demostró múltiples formaciones polipoideas de 3 a 15 mm de diámetro en el bulbo y la segunda porción duodenal. Se resecó un pólipo ulcerado con asa de diatermia mediante corriente de 10 s. El espécimen medía 2 x 2 x 2 cm. Histológicamente el pólipo mostraba ulceración del epitelio superficial y hemorragia focal (fig. 1). El pólipo estaba básicamente constituido por glándulas de Brunner hiperplásicas (fig. 2) distribuidas en la submucosa con patrón nodular a expensas de tractos conectivos fibrosos (fig. 3), con presencia de haces musculares (fig. 4). No se identificaron signos de atipia citológica ni presencia de microtrombos intracapilares. El diagnóstico fue de hamartoma de glándulas de Brunner.
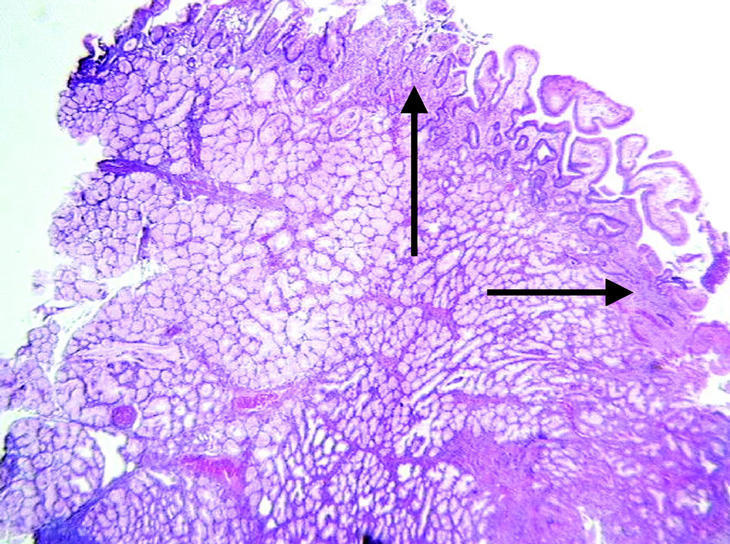
Fig. 1. Presencia en submucosa de grupos glandulares con características de glándulas de Brunner con hiperplasia marcada y patrón nodular a expensas de tractos fibromusculares. Superficie epitelial erosionada con vasos congestivos y hemorragia focal (flecha). (X40HE)
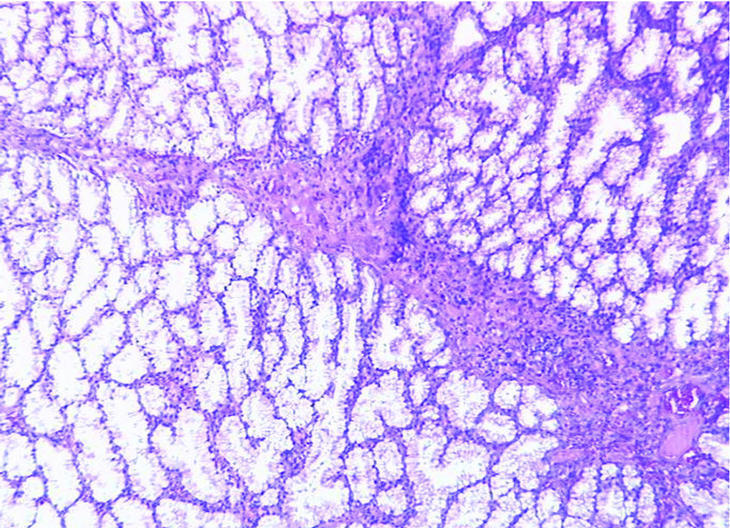
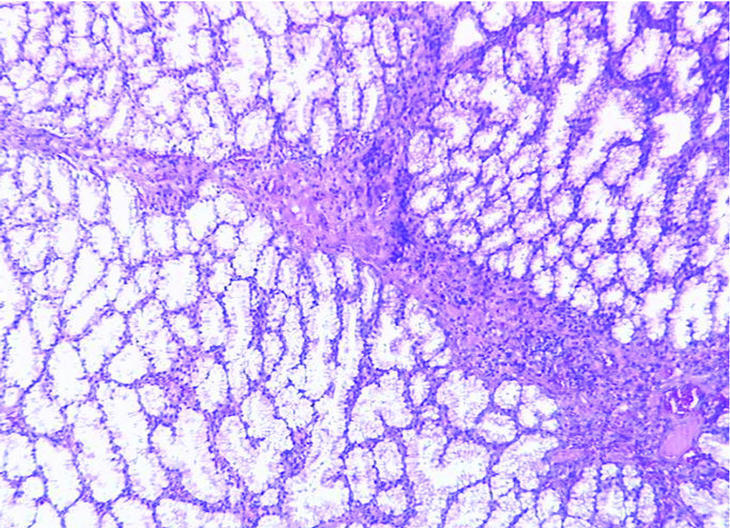
Fig. 2. Los tractos conectivos discretamente vascularizados entre las glándulas de Brunner hiperplásicas muestran moderada inflamación crónica. (X100HE).
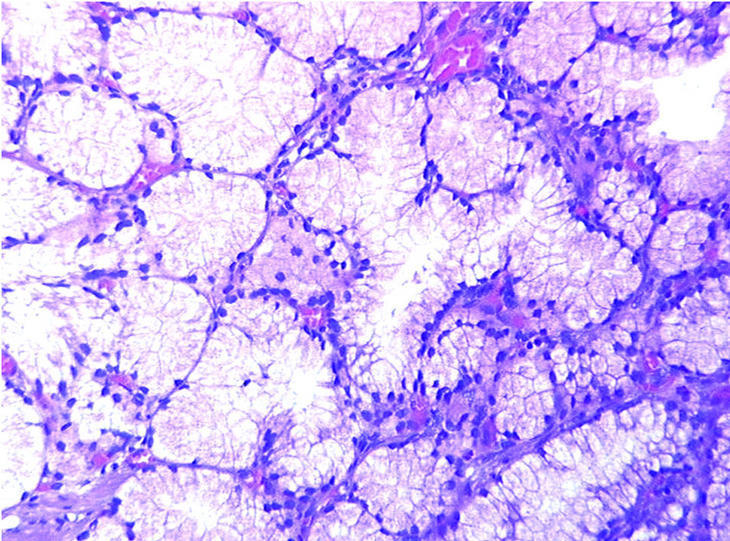
Fig. 3. A mayor aumento se observan glándulas de Brunner con citoplasma claro y núcleos redondeados, monótonas y sin atipia citológica. (X250HE).
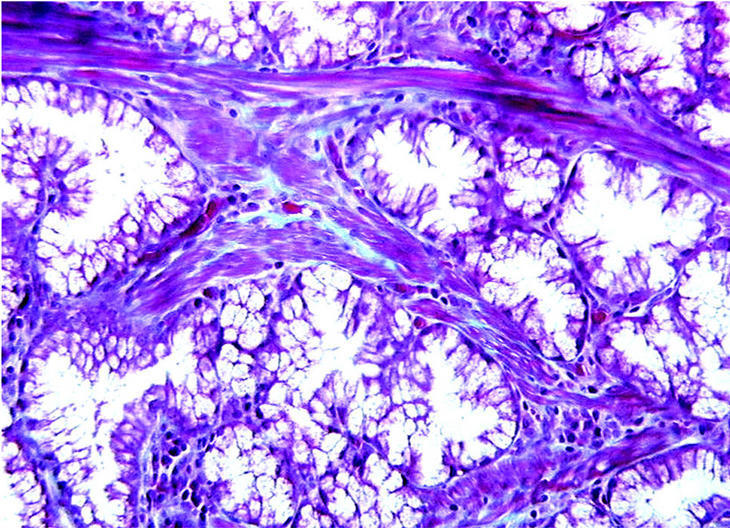
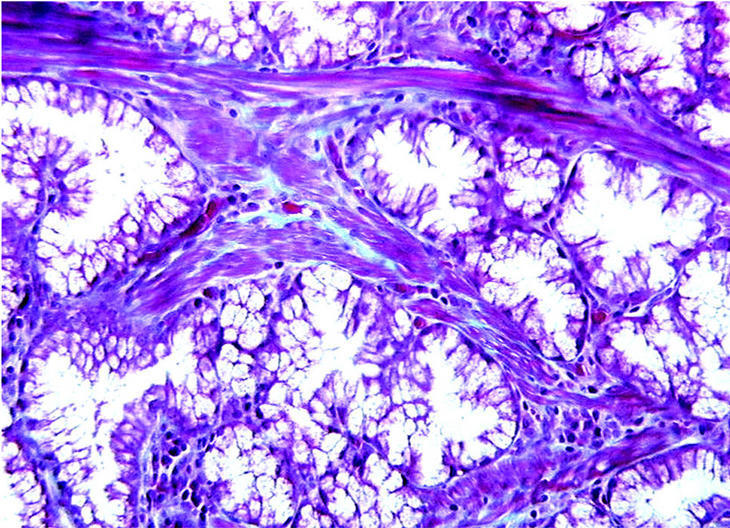
Fig. 4. Con técnica de tricrómica de Gomori se evidencia la presencia de haces musculares en los tractos conectivos que separan los grupos glandulares hiperplásicos. (X250TRICR)
Posteriormente la hemorragia permaneció inactiva con estabilidad hemodinámica y del hematócrito postransfusional del 30%. Se reinició tratamiento anticoagulante oral y el paciente permaneció estable, y se le dio el alta hospitalaria con tratamiento antisecretor (omeprazol a dosis de 20 mg/día). A los 7 meses de seguimiento no ha presentado nuevos episodios de hemorragia.
DISCUSIÓN
La HGB es una causa infrecuente de hemorragia digestiva alta1 y puede originar un espectro de lesiones localizadas fundamentalmente en el duodeno proximal. Macroscópicamente la mayoría de estos hamartomas, previamente mal llamados «adenomas» de glándulas de Brunner, son nódulos submucosos que pueden ser múltiples, bien circunscritos pero no encapsulados. Microscópicamente corresponden a proliferación nodular de glándulas de Brunner que pueden presentar ocasionales dilataciones quísticas. Entre los grupos glandulares que pueden presentar metaplasia de células de Paneth se observan tractos conectivos y haces de músculo liso, con lo que se cumplen así los criterios morfológicos para clasificarlos como lesión hamartomatosa.
La mayoría de estas lesiones son asintomáticas o se manifiestan por síntomas inespecíficos. Sin embargo, ocasionalmente las lesiones de mayor tamaño provocan complicaciones graves, principalmente hemorragia, obstrucción duodenal, intususcepción u obstrucción de la vía biliar o pancreática. Las formas de mayor tamaño (seudotumoral) son infrecuentes, pero por su tamaño pueden plantear el diagnóstico diferencial con neoplasias malignas9,10.
Estudios histológicos han encontrado una elevada prevalencia de HGB en el bulbo duodenal de pacientes con insuficiencia renal crónica11. Un estudio endoscópico demostró la existencia de hiperplasia nodular en aproximadamente un 15% de los pacientes con insuficiencia renal crónica y únicamente en un 0,3% de 300 controles sin enfermedad renal12. Sin embargo, esta relación no se ha confirmado en estudios más recientes y la mayor parte de los casos de HGB publicados son esporádicos y no relacionados con otra patología. La hipersecreción ácida gástrica observada en algunos estudios ha llevado a pensar que la sobrecarga duodenal de ácido puede desempeñar un papel en la patogenia de estos cambios duodenales1,12,13.
No existen hallazgos radiológicos o endoscópicos específicos que permitan establecer el diagnóstico diferencial con otras lesiones. Tampoco las biopsias endoscópicas superficiales lo permiten debido a su localización submucosa. El diagnóstico de certeza sólo puede establecerse mediante el examen histológico del espécimen obtenido mediante resección endoscópica o quirúrgica. Tras la confirmación histológica el tratamiento debe ser conservador, aunque la resección endoscópica o quirúrgica está indicada en los pacientes sintomáticos con complicaciones2,14,15.
La asociación de la HGB al SAP no ha sido descrita. En el SAP existe un estado de hipercoagulabilidad adquirido y la afectación gastrointestinal es infrecuente. Puede presentarse con complicaciones isquémicas agudas como trombosis de venas suprahepáticas (síndrome de Budd-Chiari), vena porta (hipertensión portal aguda) o ramas arteriales (infarto intestinal, hepático, esofágico, en vesícula biliar, colitis isquémica) o como pancreatitis6. La hemorragia digestiva es excepcional. Se ha descrito la hematemesis por varices esofágicas secundarias a trombosis de vena porta7, pero la mayoría de los casos publicados son debidos a lesiones isquémicas de la mucosa. Cappell et al8 publicaron un caso de una paciente con hemorragia digestiva en forma de melenas por úlcera de duodeno descendente refractaria al tratamiento antisecretor con antagonistas de los receptores H2 de la histamina, que requirió resección duodenal segmentaria.
En el caso que presentamos, el diagnóstico de SAP se estableció 16 años antes por criterios clínicos y de laboratorio16. Se consideró primario por la ausencia de criterios de lupus eritematoso sistémico u otra enfermedad autoinmunitaria asociada. Las complicaciones tromboembólicas pueden afectar a cualquier órgano y se caracterizan por un amplio espectro de manifestaciones clínicas según la naturaleza y el tamaño de los vasos afectados y de la cronicidad del proceso. La trombosis venosa profunda de las extremidades inferiores es la manifestación clínica más frecuente. Esta complicación se presenta hasta en un 55% de los pacientes con este síndrome tras un período de seguimiento medio inferior a 6 años. Entre éstos, más de la mitad presentará además embolia pulmonar17-19. La trombosis arterial es menos frecuente y suele manifestarse en forma de isquemia o infarto. De éstos, casi el 50% afecta al sistema nervioso central en forma de ictus o accidentes isquémicos transitorios, el 23% afecta a coronarias y el resto a diversos territorios, raramente afectados por otras causas de trombosis18. Más raros son los fenómenos embólicos por vegetaciones en la válvula mitral o aórtica17, aunque pueden demostrarse anomalías valvulares por ecocardiografía hasta en un 63% de los pacientes, en su mayoría en forma de insuficiencia valvular17.
Los anticuerpos antifosfolipídicos producen efectos procoagulantes y anticoagulantes, predominando los primeros, de ahí la clínica propia del síndrome. El efecto anticoagulante se ejerce inhibiendo la activación de los factores IX y X (poco importantes in vivo) y sobre la transformación de protrombina en trombina (vía común de la coagulación)8. El día previo a la hemorragia nuestro paciente tenía una ratio normalizada internacional de 7,2 y un TTPAr de 2,1, por lo que creemos que la alteración tan importante de las pruebas de coagulación a la llegada a la unidad de hemorragia se debió principalmente al exceso de anticoagulantes orales (en un paciente de muy difícil manejo20) y a un efecto de dilución de los factores de coagulación provocado por la intensa infusión de líquidos y hematíes que se requería para el control de una volemia efectiva, más que a un efecto del SAP. La prevención de nuevos episodios trombóticos se realiza con anticoagulantes orales a dosis altas (se ajustan para conseguir una ratio normalizada internacional entre 2 y 3)6. Evidentemente, la marcada alteración de la coagulación por el tratamiento con dicumarínicos contribuyó a la masiva hemorra-gia en este paciente.
La trombocitopenia, que también había presentado nuestro paciente, es otra manifestación prominente del síndrome, que se presentará hasta en la mitad de los casos17-19. Aunque su patogenia no se conoce bien, el tratamiento con corticoides consigue en ocasiones remisiones completas, como en el caso aquí presentado, lo que apuntaría a la participación de fenómenos autoinmunitarios o mecanismos inmunológicos21.
Recientemente se ha reconocido que algunas manifestaciones clínicas del SAP están relacionadas con el hallazgo histológico de lesiones en pequeños vasos en forma de microangiopatía trombótica6. La microangiopatía trombótica aguda por afectación de pequeños vasos es indistinguible del síndrome hemolítico urémico o de la púrpura trombótica trombocitopénica. En cambio, cuando se presenta de forma crónica conduce a un deterioro funcional más lento y progresivo del órgano afectado, y su diagnóstico puede establecerse únicamente mediante examen histológico6. Cuando afecta al riñón se presenta en forma de insuficiencia renal crónica, la mayoría de las veces asociada a hipertensión arterial22, como presentaba nuestro paciente.
En conclusión, la HGB es una posibilidad que debe considerarse en el diagnóstico diferencial de la hemorragia digestiva alta en un paciente con síndrome antifosfolipídico que esté recibiendo anticoagulantes orales y en particular en pacientes con insuficiencia renal crónica.

