




La resistencia a la insulina (RI) es un factor predictivo de respuesta al tratamiento con peginterferón y ribavirina en pacientes con hepatitis C. La RI provoca un deterioro en la sensibilidad al interferón y puede bloquear la su señalización intracelular. La RI induce la aparición de esteatosis, la progresión de la fibrosis y la liberación de citoquinas proinflamatorias al tiempo que disminuye la biodisponibilidad del interferón. Las sustancias supresoras de citoquinas 3 y las proteínas tirosín fosfatasas están implicadas en el bloqueo de la señalización intracelular del interferón y de la insulina. La RI se puede tratar mediante dieta, ejercicio físico o el uso de agentes sensibilizantes a la insulina como las biguanidas o las glitazonas. En el estudio TRIC-1 se ha demostrado que añadir metformina al tratamiento habitual mejora las posibilidades de curación en mujeres y en pacientes que consiguen normalizar la sensibilidad a la insulina durante el tratamiento.
Insulin resistance is a predictive factor of response to treatment with peginterferon and ribavirin in patients with hepatitis C. Insulin resistance impairs sensitivity to interferon and can block its intracellular signalling. Insulin resistance also induces the development of steatosis, progression of fibrosis and proinflammatory cytokine release and reduces the bioavailability of interferon. Suppressor of cytokine signalling 3 and protein tyrosine phosphatases are involved in blocking the intracellular signalling of interferon and insulin. Insulin resistance can be treated through diet, physical exercise and the use of insulin-sensitizing agents such as biguanides or glitazones. The TRIC-1 study demonstrated that adding metformin to routine treatment improves the possibilities of cure in women and in patients whose insulin sensitivity returns to normal during treatment.
La hepatitis crónica C es un problema de salud de primera magnitud. Entre 170–200 millones de personas en el mundo están infectados por el virus de la hepatitis C (VHC). Aproximadamente, el 70% de los infectados presentan datos de hepatitis crónica, y una proporción significativa de ellos desarrollarán cirrosis y hepatocarcinoma1. La historia natural de la enfermedad está influida por diferentes factores, como la edad en el momento de la infección, el sexo, consumo de alcohol, obesidad y el estado inmunológico2. En los últimos años se ha generado un gran interés científico por los disturbios metabólicos detectados en pacientes con hepatitis C, ya que son muy frecuentes y existen evidencias que soportan el papel directo del virus en su aparición. La resistencia a la insulina (RI) es un estado metabólico caracterizado por una hiperinsulinemia necesaria para mantener los niveles de glucosa dentro de la normalidad. Esta hiperinsulinemia es la responsable del daño endotelial, que condiciona las principales complicaciones de este síndrome a nivel ocular, renal, cardiaco, cerebral o hepático. Se ha implicado a la hepatitis crónica por VHC en la aparición de RI en ausencia de factores metabólicos3. Se estima que, aproximadamente, la mitad de los pacientes presentan esteatosis hepatocitaria o RI y que una cuarta parte presentan una marcada RI4,5. Los pacientes con hepatitis crónica C desarrollan diabetes mellitus (DM) tipo 2 con más frecuencia que en la población general o en pacientes con hepatitis crónica de otra etiología6. Las consecuencias de estas alteraciones metabólicas detectadas en pacientes con hepatitis C, no solo favorecen la progresión de la fibrosis, sino también afecta a la calidad de la replicación viral y a las posibilidades de curación con peginterferón y ribavirina7. El tratamiento de la RI puede constituir una nueva diana terapéutica en pacientes con hepatitis C.
Resistencia a la insulina, síndrome metabólico e infección por VHCSegún el consenso del grupo de trabajo RI de la Sociedad Española de Diabetes, la RI se define como la disminución de la capacidad de la insulina para ejercer sus acciones biológicas en tejidos diana típicos como el músculo esquelético, el hígado o el tejido adiposo8. Actualmente, se considera que la RI crónica o mantenida es la base común de numerosas enfermedades metabólicas y no metabólicas, como la DM tipo 2, la obesidad, la HTA, la enfermedad hepática por depósito de grasa, las dislipemias y/o la enfermedad cardiovascular. El síndrome metabólico se caracteriza por la presencia simultánea o secuencial de algunas de las siguientes alteraciones: RI, hiperinsulinemia compensadora, intolerancia hidrocarbonada o DM tipo 2, dislipemia aterogénica (aumento de triglicéridos VLDL, disminución del colesterol HDL), obesidad central, HTA arterial, hiperuricemia, alteraciones hemorreológicas y de la fibrinolisis (niveles elevados del inhibidor de factor activador del plasminógeno, PAI 1, estado pretrombótico), disfunción endotelial y presencia de niveles circulantes elevados de marcadores de inflamación como la proteína C reactiva. En pacientes con hepatitis por VHC genotipo 1 existen factores implicados en el desarrollo de síndrome metabólico, entre los que destacan el sobrepeso, la hiperleptinemia y la RI; a los que hay que añadir la capacidad de la proteína del core de promover RI. Esta RI puede bloquear la lipolisis y aumentar el aporte de ácidos grasos libres al hígado, al tiempo que inhibe la oxidación lipídica y el transporte, favoreciendo la formación de vacuolas grasas, así como también se acompaña de altos niveles de TNF9 y descenso en la concentración de adiponectina. Aunque la RI se detecta entre un tercio y la mitad de los pacientes con hepatitis crónica C no cirróticos no diabéticos10, la prevalencia de síndrome metabólico es baja, no superando el 5–8% de los pacientes. Por tanto, la infección por VHC modifica la señalización de la insulina y promueve resistencia, pero no ejerce el mismo efecto sobre los otros componentes que definen el síndrome metabólico.
Valoración de la sensibilidad a la insulinaLa insulina, después de unirse a su receptor, induce la fosforilación de sustratos del receptor en el hígado y músculo, e induce algunos pasos hacia la transactivación del GLUT-4, que aumenta el consumo de glucosa por las células y el depósito como glucógeno e inhibe la producción neta de glucosa por el hígado, bloqueando la glucogenolisis y neoglucogenogénesis. La insulina promueve el almacenamiento de los lípidos inhibiendo la lipólisis. Cuando la insulina no es capaz de inducir el consumo de glucosa, las células betapancreáticas incrementan la producción de insulina y el estado de hiperinsulinemia evita la hiperglucemia. De este modo, la RI depende de la secreción de insulina y de la sensibilidad a la insulina. La relación entre ambos eventos no es lineal, sino hiperbólica. La secreción de insulina aumenta cuando la sensibilidad a la insulina disminuye. Así, la concentración de insulina es usualmente alta de forma temprana en la diabetes tipo 2, pero también se ha visto en muchas personas que no son obesas o diabéticas, pero que presentan ovario poliquístico o tienen otros componentes del síndrome metabólico, como hipertrigliceridemia o HTA.
El mejor método para medir la sensibilidad a la insulina es el clamp euglucémico hiperinsulinémico. Este método mide la cantidad de glucosa requerida para mantener niveles de normoglucemia durante la infusión de insulina. Brevemente, se comienza con una perfusión de insulina de 60mU/kg/min, y cada 5min se miden los niveles de glucosa. Para evitar la hipoglucemia, la glucosa (20%) se perfunde para mantener niveles entre 90–100mg/dl. El consumo de glucosa total (valor M) depende de la glucosa infundida en los últimos 30min del test. Valores de M menores (requerimientos de glucosa menores) se han asociado con la RI. Sin embargo, el método del clamp es caro, laborioso e incómodo para su uso clínico diario.
Un modelo matemático llamado Homeostasis Model for Assessment (HOMA=insulina en ayuna [mUI/ml]×glucosa en ayunas [mmol/l]/22,5) se ha demostrado útil en la medida de la sensibilidad a la glucosa en pacientes euglucémicos. Sin embargo, esta medida no es útil en diabéticos. El HOMA no ha sido estandarizado, y aún faltan por definir los rangos de normalidad en personas sanas. En trabajos previos HOMA <2 ha sido considerado «completamente» normal y mayor de 4, como un estado prediabético. Sin embargo, estos umbrales son arbitrarios y requieren un mayor consenso.
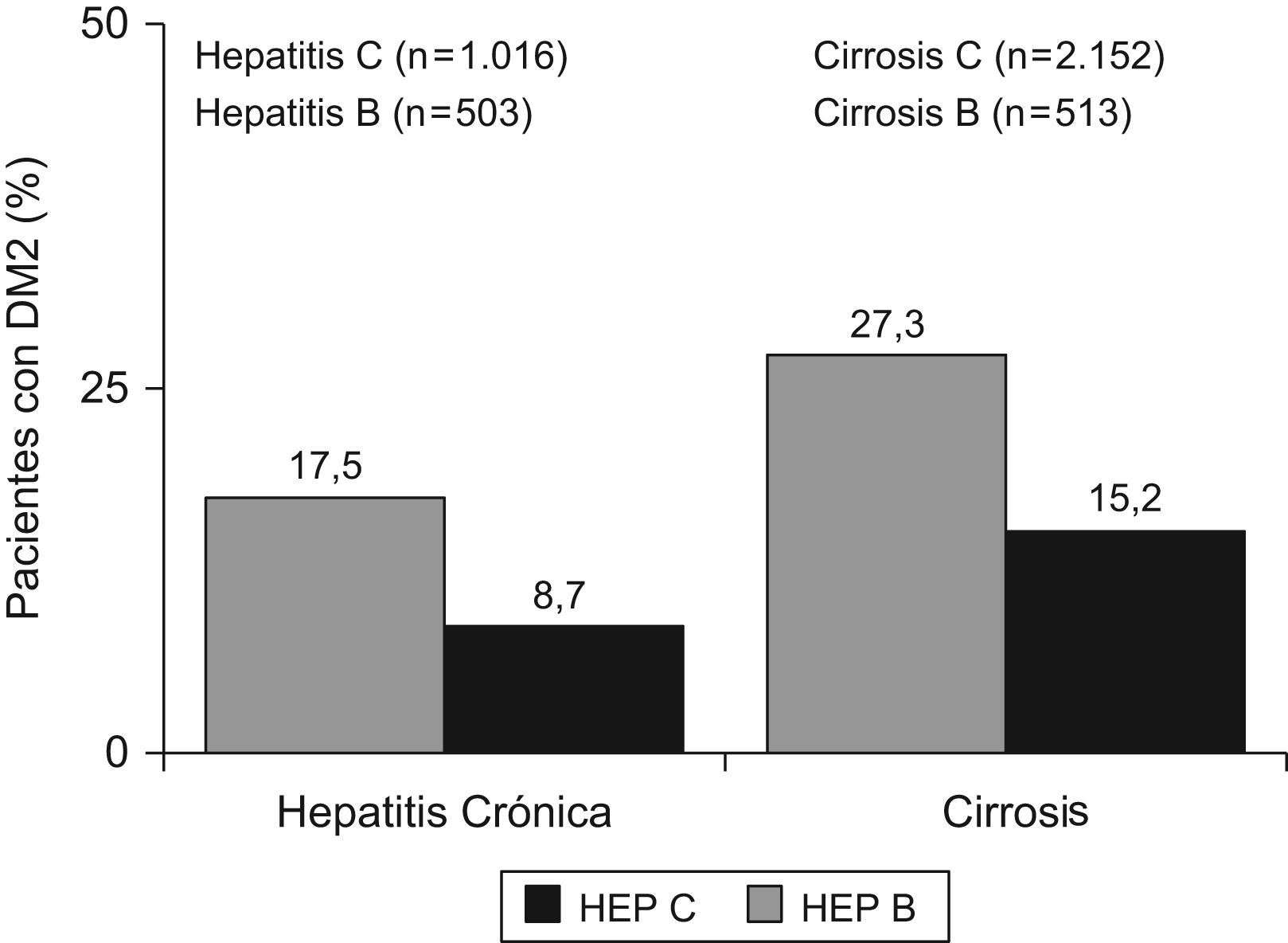
Hepatitis C y diabetesLa asociación entre la RI y la hepatitis C se restringió a los estadios más evolucionados de la enfermedad, sobre todo, en casos de fibrosis avanzada11. Sin embargo, más de la tercera parte de los pacientes presentan RI en ausencia de obesidad, diabetes o fibrosis avanzada12. La prevalencia de hepatitis C es superior en diabéticos que en controles o en pacientes con hepatitis B13. Así también, los pacientes con hepatitis crónica C presentan una tasa de diabetes que se sitúa entre el 20–50%. En un reciente metaanálisis se analizó el riesgo de diabetes en personas con hepatitis C, en comparación con la población general, y pacientes con hepatitis B14 se demostró un riesgo aumentado de padecer diabetes en los casos de hepatitis C (fig. 1). Entre 18 estudios, 15 retrospectivos y 3 prospectivos, se constató un riesgo de 1,7 veces de desarrollar diabetes en pacientes con hepatitis C. Además, en los 3 estudios prospectivos con seguimiento a largo plazo se demostró el riesgo aumentado, confirmando los hallazgos de los numerosos estudios retrospectivos. La hepatitis C aumentó el riesgo tanto en monoinfectados, en comparación con hepatitis B, como en coinfectados, en comparación con pacientes con infección VIH aislada15. En una cohorte de pacientes con hepatitis C se encontraron niveles de insulina, glucosa basal en ayunas y, por tanto, HOMA elevados, respecto a pacientes con hepatitis B, de forma que se detectó RI en el 35% de los pacientes con hepatitis C frente al 5% de los pacientes con hepatitis B, controlados por edad, fibrosis y sexo16. Además, la RI se asoció con una mayor progresión de la fibrosis, especialmente, en genotipos 1 y 4. En un estudio poblacional llevado a cabo en EE.UU. (Third National Health and Nutrition Examination Survey; NANHES-iii), que incluyó casi 10.000 pacientes, los pacientes con anti-VHC positivo mayores de 40 años presentaron un riesgo aumentado de diabetes de 3,7 veces, después de ajustar por sexo, índice de masa corporal y raza17. Por último, la asociación entre hepatitis C y diabetes se mantiene, e incluso, se incrementa en pacientes trasplantados18, más del 40% de los pacientes trasplantados por cirrosis C desarrollan diabetes en 5 años de seguimiento19.
Resistencia a la insulina como cofactor de progresión de la fibrosis hepática en hepatitis crónica por virus C
La importancia del daño hepático, en relación con la RI y la diabetes, se ha descrito extensamente en pacientes infectados por VHC. Se estableció una correlación entre la RI, la esteatosis hepática y la fibrosis en un estudio retrospectivo de 141 pacientes no diabéticos y sin tratamiento, con hígado no cirrótico, en biopsia y monoinfectados por VHC20. Otros autores también han sugerido una correlación entre la cirrosis y el riesgo de intoleracia a la glucosa o diabetes en pacientes infectados por hepatitis por VHC21,22. En pacientes infectados por VHC, con genotipo no 3, la RI y la obesidad son causas de esteatosis y por tanto acelera la progresión hacia la fibrosis23. Los posibles mecanismos, por los cuales el incremento del nivel de glucosa sérica puede contribuir a la fibrogénesis son:
- •
Hiperglucemia como resultado del incremento de la formación y depósito de los productos finales de la glicosilación, que interaccionan con receptores intrahepatocitarios, favoreciendo la colagenogénesis.
- •
Elevación de niveles de glucosa que inducen la expresión de citoquinas proinflamatorias (por ejemplo, el factor de crecimiento tisular)24.
- •
Incremento del estrés oxidativo y generación de radicales libres que activan una respuesta celular ante el estrés, que es llave de liberación de citoquinas proinflamatorias (como TNF α e IL-6)
Por tanto, la RI se asocia a una mayor progresión de la fibrosis25, probablemente, por su implicación en el desarrollo de esteatosis hepática, que provoca una mayor actividad necroinflamatoria y acelera la progresión de la fibrosis26,27. Durante el proceso de la replicación, las proteínas virales son agrupadas y plegadas correctamente por las chaperonas en el retículo endoplasmático (RE), pero, en algunas circunstancias, el RE falla al exportar las proteínas adecuadamente sintetizadas y tiene lugar una acumulación de proteínas incorrectamente plegadas. Estas proteínas defectuosas causan la disfunción del RE y promueven la inflamación y el estrés oxidativo, lo que condiciona la progresión de la fibrosis28. Entre otros factores implicados en la progresión de la fibrosis en pacientes con hepatitis C y RI están: a) la esteatosis29; b) hipoadiponectinemia30; c) hiperleptinemia31, y d) incremento de los niveles de TNF.
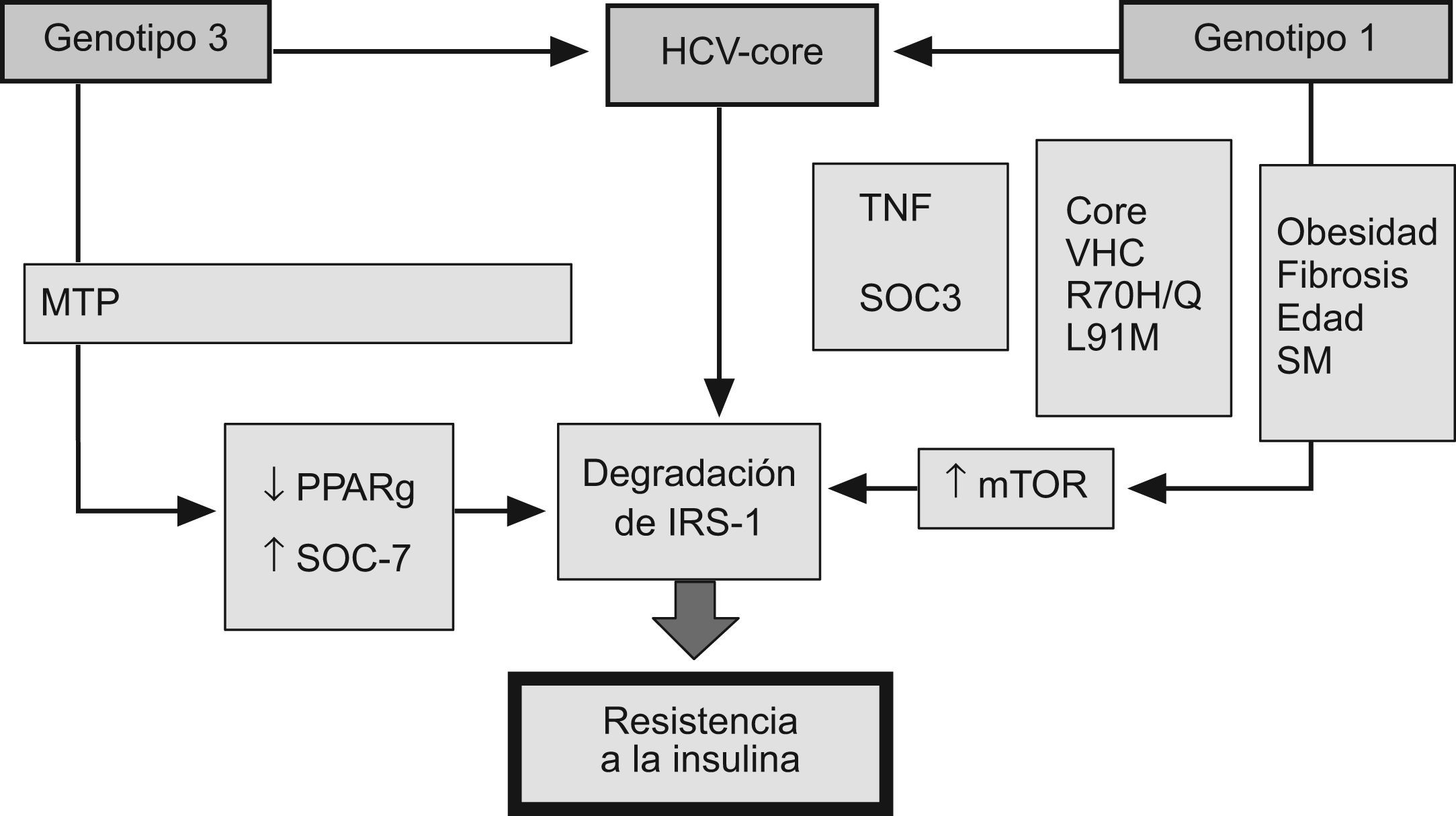
Mecanismos moleculares de la resistencia a la insulina en la hepatitis CLa proteína del core del VHC ha sido implicada en el proceso de inducción a la resistencia de la insulina. Los ratones transgénicos que expresan esta proteína desarrollan RI, lo que no ocurre en ratones no modificados genéticamente32. Durante la replicación del VHC, la proteína del core contacta con la membrana exterior de la mitocondria y, posteriormente, se ensambla en el RE33. Este proceso es especialmente eficiente en casos de esteatosis intracitoplásmica, lo que hace que la replicación viral sea mayor en pacientes con RI y esteatosis. La proteína del core inhibe la expresión del peroxisome proliferator-activated receptor (PPAR) α y γ, e induce la sobreproducción de TNF α, lo que promueve la degradación del receptor de la insulina-1 (IRS-1)34. Proteínas no estructurales, como la NS3 y NS5, interactúan con el RE. La NS3 realza la actividad de la NADPH oxidasa 2 (NOX2) e incrementa las proteínas nitrosiladas y los radicales libres. Las proteínas NS5A y NS5B activan el receptor toll like 4 y la vía NFkB, incrementando el TNF y la prodcción de IL-6, lo que finalmente promueve la RI35. La proteína del core del VHC interfiere con las señales intracelulares de la insulina por mecanismos dependientes del genotipo viral (fig. 5)36,37. En el genotipo 1, la proteína del core aumenta la síntesis del receptor de la rapamicina en mamíferos (mTOR), contribuyendo con ello al mantenimiento de unos niveles elevados de replicación viral y desarrollo de RI. Por el contrario, en cultivos celulares transfectados por VHC genotipo3,38, la RI se debe al incremento de la producción de la señal de la sustancia supresora de citocinas (SOC) y la disminución de la expresión de PPAR γ39,40. De hecho, las SOC inhiben la fosforilación de Akt y fosfatidil inositol 3 kinasa (PI3K), bloqueando con ello la señalización intracelular de la insulina e inhibiendo la transactivación del GLUT-4, lo que evita la captación de glucosa por las células41 y favorece el desarrollo de RI. Desde el descubrimiento en 1989 del VHC, su propagación en cultivos celulares ha sido el objetivo de mayor importancia para virólogos de todo el mundo. Los virus que causan la hepatitis son muy difíciles de mantener en cultivos celulares y, de hecho, este sistema no está disponible actualmente para el virus de la hepatitis B (VHB). Este problema también está vinculado a la naturaleza de la célula huésped, debido a que los hepatocitos humanos diferenciados son muy difíciles de propagar en cultivo celular. Sin embargo, se ha demostrado que la sangre o suero de pacientes con infección crónica del VHC es infecciosa in vivo, ya que contiene viriones infecciosos aptos para la infección in vitro de hepatocitos en cultivo. Las células derivadas de hepatoma humano Huh7 y líneas de células HepG2 podrían emplearse para la replicación del VHC, al igual que otras líneas de células hepáticas inmortalizadas por el antígeno SV-40 T42. Ito et al43 han realizado cultivos de hepatocitos primarios de pacientes con infección crónica por VHC, obteniendo títulos virales relativamente altos en las células y en el sobrenadante. Los hepatocitos primarios de humanos y chimpancés son susceptibles de infección por el VHC durante un tiempo muy limitado, durante el cual estas células son utilizables como modelo experimental44,45.
Los replicones son construcciones genómicas o subgenómicas que expresan el complejo enzimático para la proliferación de virus, siendo capaces de una replicación viral autónoma. Estos replicones han resultado ser una herramienta de gran utilidad para el estudio de la replicación del VHC y para analizar los efectos de distintos antivirales contra las principales enzimas diana (ej.: la proteasa NS3/4 y la polimerasa NS5B). El replicón JFH-1, desarrollado por el grupo de Takaji Wakita y aislado de un paciente japonés con hepatitis fulminante46, es el primer clon auténtico de VHC (con genotipo 2b) considerado por la comunidad científica como capaz de crecer en cultivo celular. La transfección del genoma completo del JFH-1 en la línea celular Huh7 conduce a la producción de partículas de VHC capaces de infectar células naive y a chimpancés. También se ha descrito un sistema similar para el genotipo 1a (H77) para la producción de virus, aunque con menor titulación47. Por otra parte, se ha desarrollado un método alternativo de infección para hepatocitos humanos en cultivo48, dado que las células Huh7 no pueden ser infectadas con partículas de VHC obtenidas de pacientes infectados. La infección por VHC se caracteriza por una alta tasa de progresión a fibrosis, hepatitis crónica, aparición de cirrosis y, en último término, carcinoma hepatocelular. Diferentes observaciones sugieren que la esteatosis hepática es una característica histológica común en la infección crónica por VHC. Además, existen numerosas evidencias que apuntan a que la esteatosis hepática como un factor de riesgo para el desarrollo de cirrosis. Esto sugiere que el VHC tiene un papel directo en el desarrollo de la esteatosis y/o que la presencia de esteatosis afecta a la progresión de enfermedades hepáticas relacionadas con el VHC. La proteína del core es un componente del VHC, cuya implicación en la esteatosis, fibrosis y carcinogénesis hepática es bien conocida49,50. Algunos estudios sugieren que la proteína del core provoca la aparición de esteatosis a través de la inhibición de la actividad de la proteína de transporte de triglicéridos de microsomas (MTP) y la secreción de VLDL y el deterioro de la expresión y actividad transcripcional de PPAR α51. La proteína sterol regulatory element binding protein 1s (SREBP1s) es un miembro de la familia de factores de transcripción denominada de cremallera de leucina hélice-loop-hélice. El papel fundamental de las 2 isoformas SREBP1 es la regulación de la síntesis de ácidos grasos en el hígado, conocido por los estudios en ratones transgénicos que sobreexpresan constitutivamente las forma madura de SREBP152. Estos estudios sugieren que SREBP1s incrementa la transcripción de genes implicados en la síntesis hepática de ácidos grasos hepáticos (entre ellos ácido graso sintasa [FAS], ácido graso sintasa, acetil-CoA carboxilasa [ACC], estearoil-CoA desaturasa [SCD]), induciendo esteatosis hepática masiva a través de una mayor acumulación de triglicéridos. La proteína PPAR γ es un factor de transcripción que pertenece a la superfamilia de proteínas receptores nucleares. Es el regulador principal de la diferenciación de los adipocitos y juega un papel crucial en la regulación de una serie de genes implicados en el metabolismo de la glucosa y los ácidos grasos53. Hay evidencias que indican que la proteína PPAR γ en el hígado incrementa la trascripción de ciertos genes responsables de la síntesis de ácidos grasos hepáticos (incluyendo FAS, ACC, SCD), así como de otros genes implicados en la absorción de ácidos grasos (como FAT/CD36, un transportador de ácidos grasos). Por lo tanto, la proteína PPAR γ en el hígado contribuye a la regulación de la síntesis de lípidos, el transporte y almacenamiento de los mismos en los hepatocitos, causando así el desarrollo de la esteatosis hepática. Por otra parte, Kim et al54, utilizando un modelo basado en cultivo celular, han demostrado que la proteína del core del VHC es capaz de inducir la expresión génica y actividad transcripcional de SREBP1, causando así el aumento de la síntesis de ácidos grasos. También observaron que la proteína del core del VHC eleva la actividad de PPAR γ, induciendo la expresión de los genes asociados a la absorción de ácidos grasos. Estos resultados sugieren que SREBP1 y PPAR γ pueden representar una nueva diana terapéutica potencial en la patogénesis de la infección por el VHC. La patogenia de la esteatosis se debe tanto a factores virales como del huésped. La esteatosis viral se da, sobre todo, en pacientes infectados con virus del genotipo 3a, en los que la acumulación de grasa se correlaciona con el nivel de la replicación del VHC en el suero y en el hígado, y desaparece después del tratamiento antiviral, lo que indica una función directa de productos específicos del virus en la deposición de grasa intracelular. Para abordar el papel del genotipo específico del VHC sobre la acumulación de lípidos en las células, Abid et al55 desarrollaron un modelo in vitro para estudiar el efecto de la proteína del core perteneciente a varios genotipos virales (1b, 2a, 3a, 3h, 4h y 5a). Llegaron a la conclusión de que el patrón observado en las células Huh7 sobre la expresión de las 6 proteínas del core corroboraba, en gran medida, el fenotipo observado in vivo. La proteína del core derivada del genotipo 3a era unas tres veces más eficiente en la inducción de la acumulación de triglicéridos en las células transfectadas que la proteína correspondiente del genotipo 1b, y se postula como el candidato ideal para estudiar la patogenia de la esteatosis inducida por hepatitis C. Este grupo también estudió la función de la expresión de PPAR γ en la acumulación de triglicéridos en células Huh7 transfectadas con las proteínas del core de los genotipos 1b y 3a24. Demostraron que la expresión de la proteína del core del VHC genotipo 3a correlacionaba con un aumento en la acumulación de triglicéridos y con una reducción significativa de los niveles de mRNA de PPAR γ, en comparación con el genotipo 1b. Por otra parte, Fukasawa et al56 demostraron que la ACC1 y la FAS, enzimas responsables de la biosíntesis de novo de lípidos, se inducen en células Huh7 transfectadas con la proteína del core del VHC. Utilizando el análisis de DNA-microarray, Pazienza et al57 compararon el perfil de expresión génica de células Huh7 transfectadas con la proteína del core del VHC de los genotipos 1b y 3a. Concluyeron que la expresión de diversos genes relacionados con el transporte y el metabolismo de lípidos se inducen o se reprimen de manera genotipo dependiente. Este hecho podría explicar distintos grados de esteatosis asociada a la infección por VHC. La morbilidad asociada con el VHC se debe también a distintas manifestaciones extrahepáticas. Entre ellas, la diabetes ha sido el foco de mayor atención en la investigación. Existen evidencias epidemiológicas que vinculan estrechamente la infección por VHC y la diabetes. Los pacientes con hepatitis C crónica son más propensos a sufrir diabetes tipo 258 y los pacientes diabéticos son más propensos a ser infectados por el VHC. Aunque la RI aumenta en paralelo con la edad y el estadio de fibrosis hepática, es significativamente mayor en las primeras etapas de la hepatitis C crónica59, lo que sugiere un papel directo del VHC en la homeostasis de la glucosa. Pazienza et al emplearon el modelo anteriormente descrito de la expresión transitoria de la proteína del core de los diferentes genotipos del VHC para evaluar la interacción de la proteína del genotipo 3a del VHC, con la vía de señalización de la insulina, comparándola con la del genotipo 1b. Demostraron que los niveles de proteína del IRS-1 se redujeron significativamente en células Huh-7 que expresaban las proteínas del core de ambos genotipos 3a y 1b. Sin embargo, mientras la proteína del core del genotipo 1b promueve la degradación de IRS-1 mediante la inhibición de PPAR γ e inducción de suppressor of cytokine signal-7 (SOCS-7), la proteína del core del genotipo 1b activaba mTOR, demostrando así la existencia de mecanismos diferentes para proteínas genéticamente distintas. Todavía quedan muchos mecanismos moleculares por dilucidar sobre la interacción VHC-huésped que permitan el desarrollo de medicamentos eficaces para el tratamiento de esta enfermedad. En este sentido, los sistemas de replicación viral basados en cultivo celular descritos hasta ahora serán de gran ayuda en esta tarea. La disponibilidad de los sistemas para la replicación de todos los genotipos del VHC conocidos es muy deseable, a fin de averiguar la importancia del genoma del virus en el desarrollo de la infección y patologías asociadas.
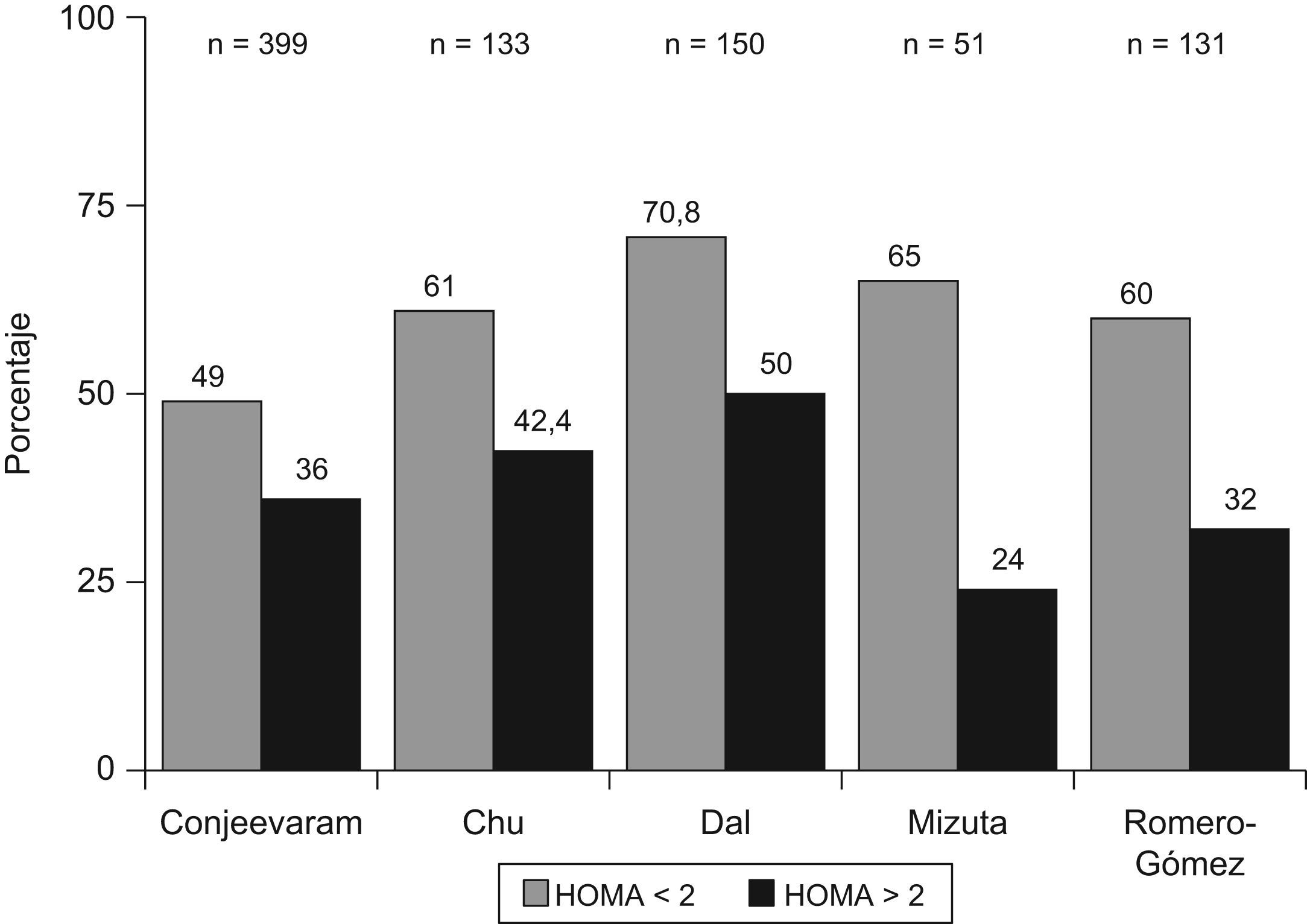
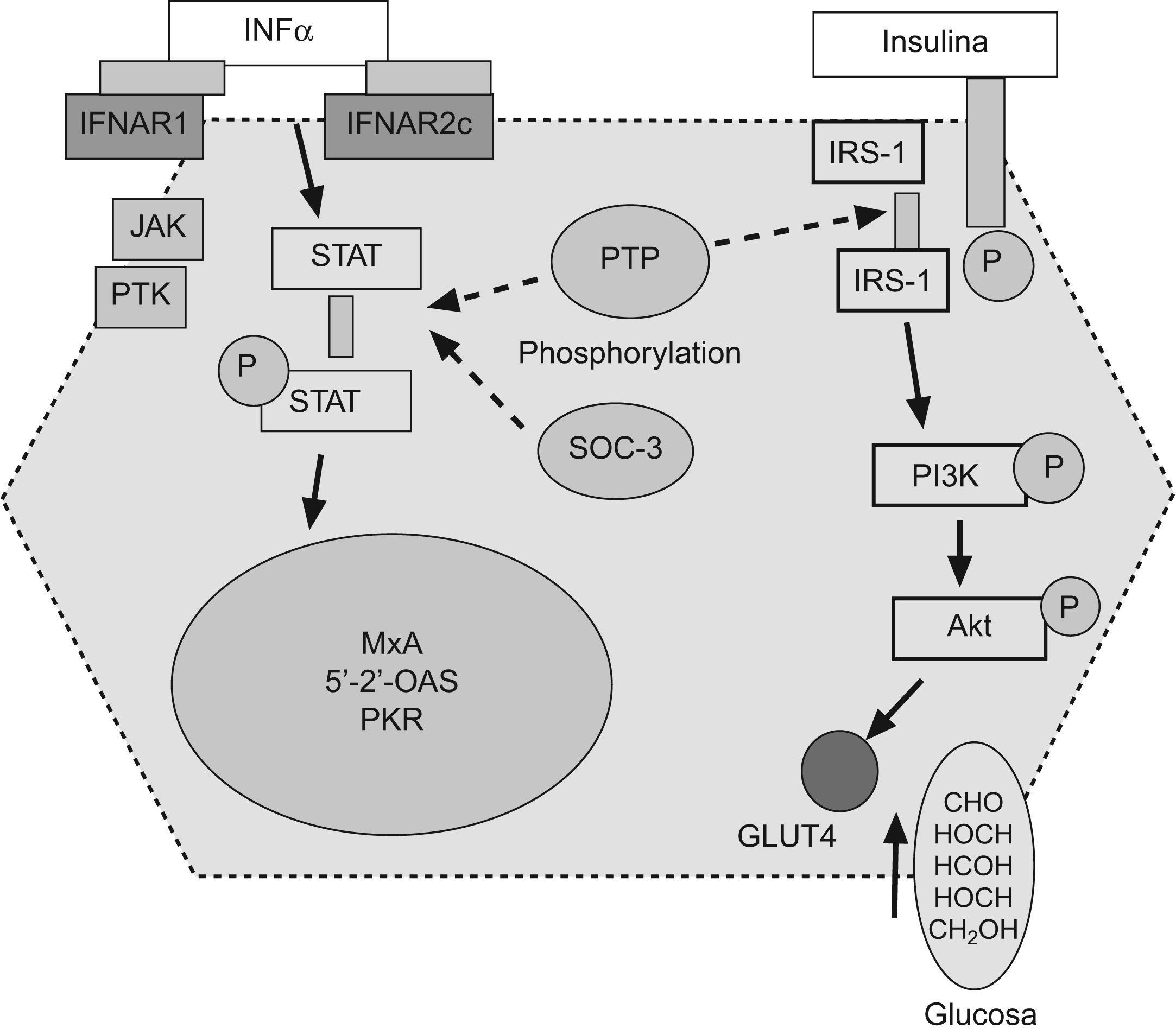
Resistencia a la insulina y respuesta a la terapia antiviral en hepatitis crónica CLos factores asociados a la no respuesta al tratamiento antiviral en pacientes infectados por el VHC son la carga viral, la fibrosis avanzada, el genotipo viral (tipo 1) y la RI. En pacientes con hepatitis C genotipo 1, la presencia de RI se asocia a una menor y significativa tasa de respuesta viral sostenida tanto en pacientes europeos12, americanos60 o asiáticos61,62. (fig. 2). En 131 pacientes españoles con hepatitis C genotipo 1 tratados con peginterferón y ribavirina durante un año, la tasa de respuesta viral sostenida fue del 60% en pacientes con HOMA<2; del 40% en pacientes con HOMA entre 2–4, y de solo el 20% en pacientes con HOMA>4 (fig. 2). No obstante, algunos estudios no encuentran dicha asociación o la RI no llega a ser una variable independiente en la predicción de respuesta. Los motivos por los que ocurren estas discrepancias no están establecidos, pero podrían incluir la extrema colinealidad entre las variables incluidas en los estudios. En el trabajo de Walsh63, la variable independiente fue la obesidad, mientras que en la cohorte de genotipo 1 no diabéticos de Camma64 la variable independiente fue la esteatosis. La esteatosis, obesidad, resistencia a la (fig. 3) insulina, los niveles de GGT y la edad estan estrechamente relacionados y definen situaciones metabólicas, en gran parte, superponibles, lo que hace que cualquiera de ellas pueda emerger como variable independiente y desplazar a las demás. Por otro lado, la RI se valora por un método indirecto, el cálculo del HOMA, que consiste en calcularlo a partir de la glucemia e insulinemia en ayunas. Por tanto, las condiciones de extracción de muestras en ayunas rigurosa, la secreción de la insulina en pulsos por parte del páncreas y el consumo de glucosa, que tiene lugar en muestras congeladas por largos periodos de tiempo, son factores que inciden en el resultado final y pueden generar falsos positivos (ausencia de ayunas) o negativos (muestras congeladas con consumo de glucosa). Además, los métodos utilizados en la determinación de la insulinemia no están estandarizados y no siempre son comparables los resultados entre laboratorios, por lo que se requiere una medición centralizada o un control preanalisis65. La asociación RI y respuesta viral se ha constatado en pacientes con genotipo 1 y en pacientes con genotipo 3, pero los estudios que incluyen mezclas de pacientes de diferentes genotipos pueden no detectar diferencias. La diversidad viral es amplia y, recientemente, se ha descrito que la sustitución aminoacídica en la posición 70 y/o 91 condiciona una proteína del core capaz de promover al mismo tiempo una RI marcada y una menor tasa de respuesta al tratamiento antiviral66. Por tanto, la distribución de virus portadores de estos cambios aminoacídicos en la proteína del core podría ser otro factor de confusión. Por tanto, la relación RI y pérdida de respuesta viral sostenida podría deberse a 2 situaciones diferentes. Por un lado, ese disturbio metabólico es el común denominador de todas las situaciones relacionadas con la no respuesta, como la obesidad, la edad, la raza afroamericana o la fibrosis avanzada. Pero por otro lado, el VHC promueve el desarrollo de RI y este deterioro de la señalización intracelular de la insulina podría condicionar la actividad antiviral del interferón. En medios de cultivos celulares transfectados por VHC, la adición de insulina junto al interferón inhibe el efecto antiviral de este y disminuye la síntesis de proteínas antivirales, como PKR, además, parece que el interferón requiere una señalización normal a la insulina para su correcto funcionamiento67. La tirosín-fosfatasa (PTP) puede bloquear la señalización intracelular de la insulina al inhibir la fosforilación del sustrato del IRS-1 y al mismo tiempo, inhibir la fosforilación de las signal transcription activation (STAT) evento crucial en la inhibición de la replicación viral. Además, en situación de RI (por ejemplo, tras silenciar IRS-1 con RNAi) aumenta la actividad de las PTP lo que generaría una mayor resistencia al interferón68.Por otro lado, la infección viral aumenta los niveles de SOC como SOC-3, implicada tanto en la aparición de RI como en el deterioro de la señalización intracelular del interferón.
 en pacientes con resistencia a la insulina (HOMA>2), que oscila entre un 13–41%.")

Las sustancias supresoras de citoquinas y las proteínas tirosin-fosfatasas podrían interferir tanto la señalización intracelular de la insulina como del interferón. IFNa: interferón α, IFNAR1: receptor-1 del interferón, JAK: janus kinasa, PTK: proteina tirosina kinasa, PKR: proteína kinasa R, IRS-1: receptor soluble-1 de la insulina, PI3K: fosfatidil-inositol-3-kinasa.
Por tanto, la RI parece estar implicada en la disminución de la sensibilidad al interferón, y el bloqueo de estas vías moleculares podría conformar una nueva diana terapéutica en el manejo de pacientes difíciles de curar.
Desde un punto de vista clínico, existen numerosas evidencias que conectan directamente la replicación viral activa con la aparición de RI y sobre todo, la desaparición o mejora de este disturbio metabólico, una vez eliminado el virus. En pacientes con aclaramiento viral sostenido se comprobó un descenso de la tasa de RI, al tiempo que en no respondedores no se apreciaban modificaciones. En una cohorte de 1.059 pacientes con hepatitis crónica C, los pacientes se clasificaron según presentasen una glucemia plasmática basal normal, una hiperglucemia no diabética y una diabetes. Todos los pacientes recibieron un tratamiento combinado con interferón pegilado y ribavirina durante 24 o 48 semanas, en función del genotipo viral. Se constató el efecto negativo de la glucemia basal alterada (glucemia >100mg/dl) en la tasa de respuesta viral sostenida. Además, en los pacientes con glucemia basal normal, la tasa de desarrollo de glucemia basal alterada o DM tipo 2 fue significativamente menor en pacientes que alcanzaron respuesta viral sostenida, en comparación con los no respondedores69. Además, se ha demostrado que la normalización del HOMA en pacientes con respuesta sostenida se acompañó de un aumento en la expresión de IRS-1 en el hígado mediante estudios de inmunohistoquímica, lo que avala que la erradicación del VHC permite recuperar la sensibilidad a la insulina, al menos, en la parte de alteración debida directamente a la acción del virus.
Tratamiento de la resistencia a la insulina en pacientes con hepatitis crónica CLa RI puede tratarse mediante ejercicio físico, dieta o el uso de fármacos que mejoran la sensibilidad a la insulina. Los fármacos sensibilizantes disponibles en el mercado son las biguanidas (metformina) y las glitazonas (pioglitazona y rosiglitazona). Existen comunicaciones aisladas que demuestran que una intervención en el estilo de vida eliminando el sedentarismo y promoviendo una dieta mediterránea ejerce un efecto beneficioso y podría mejorar las posibilidades de curación de pacientes con hepatitis C. En 32 pacientes con genotipo 1 y síndrome metabólico sometidos a dieta y ejercicio físico durante 12 semanas se comprobó una mayor tasa de respuesta viral sostenida tras tratamiento con peginterferón y ribavirina durante 48 semanas, en aquellos que consiguieron mejorar el índice de RI70.
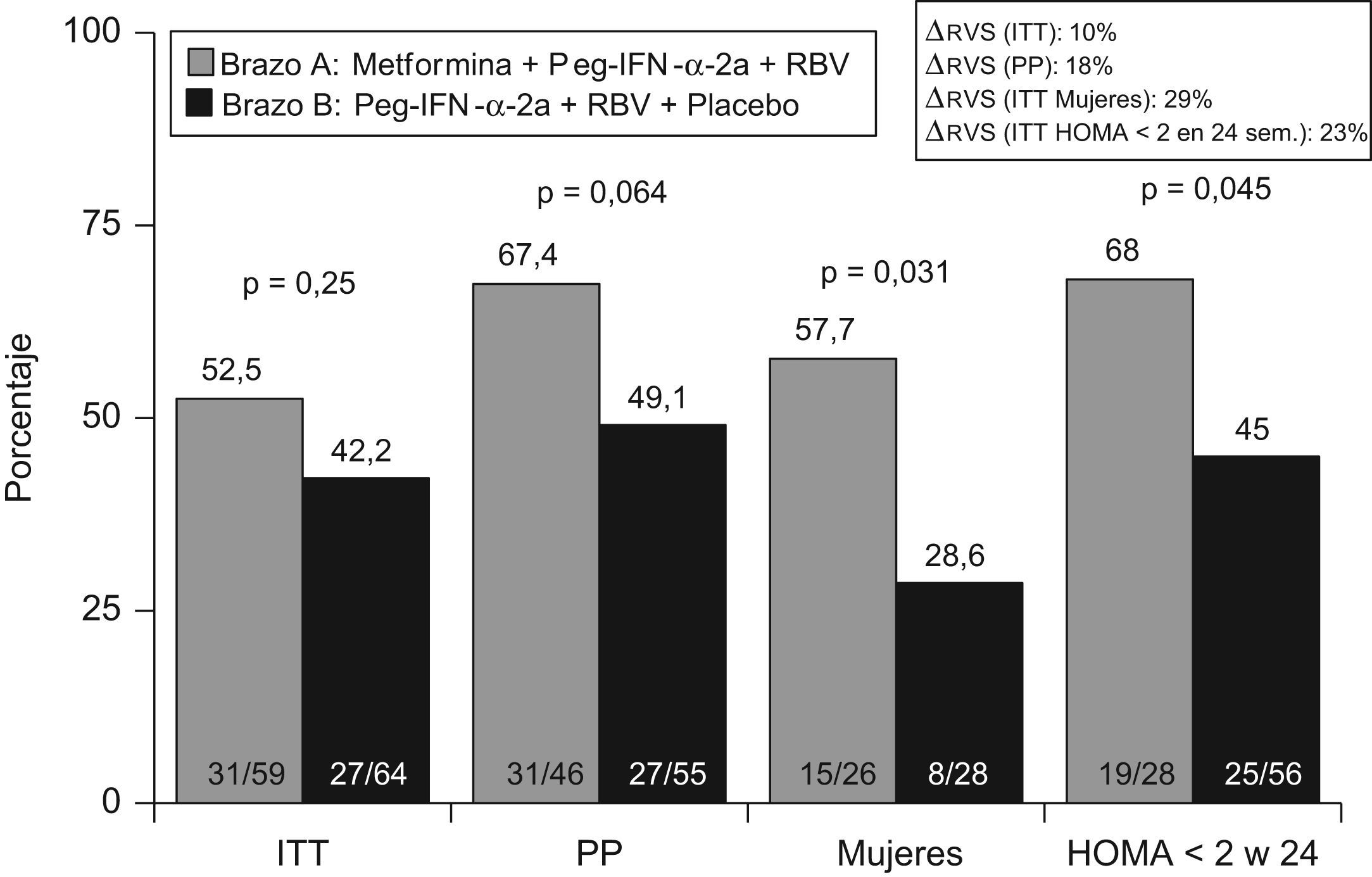
Recientemente, en un ensayo clínico (TRIC-1) controlado, aleatorizado y doble ciego, se ha analizado el efecto de la adición de metformina a la terapia estándar en el tratamiento de la hepatitis C (fig. 4). En una cohorte de 123 pacientes, con hepatitis C genotipo 1 y RI se administró metformina o placebo de forma concomitante a peginterferón α y ribavirina. En el análisis por intención de tratar la metformina mejoró la tasa de respuesta un 10% (52 vs. 42%; p=ns). Así también, más de la mitad de los pacientes en (fig. 5) tratamiento con metformina alcanzó un HOMA<2 a las 24 semanas de tratamiento, y este evento se asoció con las posibilidades de alcanzar respuesta sostenida a las 24 semanas de seguimiento. Por otro lado, las mujeres que recibieron metformina doblaron las posibilidades de curación, en comparación con el grupo que recibió placebo (57 vs. 28%; p=0,03). Los mecanismos por los que la metformina podría mejorar la sensibilidad al interferón y ribavirina son desconocidos. No obstante, en adolescentes se ha comprobado que la metformina induce una mayor pérdida de peso en mujeres que en varones. Por otro lado, el estudio TRIC-1 se demostró que las mujeres con metformina presentaron una mayor caída de la carga viral durante las 12 primeras semanas, significativamente superior a las mujeres sin metformina y estas diferencias no se observaron en los hombres. Por último, metformina fue bien tolerada, salvo por el desarrollo de diarrea en más de un tercio de los casos, aunque en ninguno de ellos fue necesario suspender la medicación. Dos pacientes desarrollaron hepatitis colestásica y no modificó los niveles de lactato monitorizados durante el tratamiento. Por lo tanto, añadir metformina al tratamiento antiviral con peginterferón y ribavirina es seguro, mejora la RI y en mujeres puede doblar las posibilidades de curación.
. El tratamiento con metformina, peginterferón y ribavirina duplicó la respuesta viral sostenida en mujeres y en personas que normalizaron el indice de HOMA a las 24 semanas de tratamiento.")
En el ensayo clínico TRIC-1 se ha analizado el efecto de añadir metformina a la terapia estándar en el tratamiento de la hepatitis C, siendo un fármaco seguro y bien tolerado. El análisis por ITT demostró que añadir metformina mejoró en un 10% la tasa de respuesta viral sostenida (42 vs. 52%; p=ns). El tratamiento con metformina, peginterferón y ribavirina duplicó la respuesta viral sostenida en mujeres y en personas que normalizaron el indice de HOMA a las 24 semanas de tratamiento.
 favoreciendo la degradación del receptor soluble de insulina. Por el contrario, la resistencia a la insulina en el genotipo 3 se debe al incremento de la producción de la señal de la sustancia supresora de citocinas (SOC) y la disminución de la expresión de PPAR γ.")
El mecanismo implicado en el desarrollo de resistencia a la insulina en la infección por virus C depende del genotipo viral. En el genotipo 1, la proteína del core aumenta la síntesis del receptor de la rapamicina en mamíferos (mTOR) favoreciendo la degradación del receptor soluble de insulina. Por el contrario, la resistencia a la insulina en el genotipo 3 se debe al incremento de la producción de la señal de la sustancia supresora de citocinas (SOC) y la disminución de la expresión de PPAR γ.
La pioglitazona se ha utilizado en el tratamiento de la RI en pacientes con hepatitis C. En un estudio que incluyó a 40 pacientes genotipo 1 con RI que se aleatorizaron a recibir pioglitazona o placebo, junto a la terapia con peginterferón y ribavirina. La respuesta al final del tratamiento fue superior en el grupo de pioglitazona pero la tasa de respuesta viral sostenida fue la misma en ambos brazos71. El escaso número de pacientes no permite alcanzar conclusiones definitivas. Por otro lado, en un estudio secuencial, que incluyó 30 pacientes distribuidos en 3 grupos, delgados sin tratamiento y obesos, con y sin tratamiento. El tratamiento se inició 4 semanas antes de la terapia antiviral. La respuesta viral rápida a las 4 semanas de tratamiento fue superior en pacientes obesos que recibían pioglitazona en comparación con los pacientes obesos que recibieron placebo72.
Por último, en casos aislados, el tratamiento secuencial parece ser superior al tratamiento concomitante. En un paciente no respondedor genotipo 3 el uso previo de pioglitazona a dosis altas (45mg/d) se acompañó de respuesta viral sostenida. En cambio, en 5 pacientes previamente no respondedores infectados por diferentes genotipos la triple terapia concomitante no consiguió el aclaramiento viral en ningún caso73. No obstante, es necesario diseñar nuevos esudios, incluyendo un número suficiente de casos y aleatorizados, según genotipo, para definir la estrategia terapéutica óptima en el manejo de la RI en pacientes con hepatitis C.
ConclusionesLos pacientes con hepatitis crónica C presentan mayor RI que los pacientes con hepatopatías víricas (no hepatitis C) y metabólicas, a pesar de una distribución similar por sexo, índice de masa corporal, antecedentes familiares de diabetes y estadio de fibrosis. La RI se relaciona con el nivel del ARN del VHC y es mayor en pacientes con hepatitis C que en controles sanos74. El VHC promueve el desarrollo de RI. La proteína del core induce la degradación del sustrato del receptor de la insulina por un mecanismo dependiente de genotipo. El estado de pérdida de sensibilidad a la insulina favorece la replicación viral, aumenta la progresión de la fibrosis y se asocia a menor posibilidad de curación. La erradicación del virus durante y después del tratamiento se acompaña de un descenso de la RI y, posteriormente, a una caída de la tasa de glucemia basal alterada y DM tipo 2. La RI disminuye las posibilidades de curación. El tratamiento de la RI con metformina parece prometedor en mujeres con genotipo 1 y HOMA>2, mientras que los datos comunicados con pioglitazona son desesperanzadores, aunque la selección de pacientes con genotipo 3 y el uso secuencial podría mejorar los resultados y debería testarse en futuros estudios.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

