





INTRODUCCIÓN
El desarrollo de la fibrosis hepática es un proceso dinámico que provoca la interrupción de las funciones hepáticas con disfunción del órgano. Generalmente, la fibrosis hepática se produce como una respuesta a la lesión hepatocelular crónica provocada por el abuso de bebidas alcohólicas, infecciones crónicas, enfermedades biliares y afecciones congénitas metabólicas, entre otras, y puede llegar a conducir a largo plazo al desarrollo de cirrosis y carcinoma hepatocelular.
Aunque el proceso de la fibrosis hepática aún no se conoce totalmente, durante los últimos años los continuos estudios acerca de su biología celular y molecular han permitido comprender muchos detalles relacionados con su fisiopatogenia. La identificación de las células estrelladas del hígado (también llamadas lipocitos, células almacenadoras de grasa o células de Ito) y su gran importancia en la fibrosis hepática no solamente han modificado el enfoque de las investigaciones sobre la base patogénica de la fibrosis hepática, sino que también han abierto nuevas líneas de interesantes investigaciones sobre futuras estrategias terapéuticas capaces de frenar el proceso fibrogénico.
CONCEPTOS GENERALES SOBRE LAS CÉLULAS ESTRELLADAS
Fue Karl Kupffer quien en el año 1876 descubrió las células estrelladas del hígado (CEH) cuando intentaba buscar fibras nerviosas en el hígado utilizando la técnica de cloruro de oro1. Logró así identificar un grupo de células que presentaban prolongaciones que entraban en contacto con la pared sinusoidal y con los hepatocitos, a las que llamó «Sternzellen» (células estrelladas).
Hoy sabemos que las células estrelladas son células mesenquimales, perisinusoidales ubicadas a lo largo de los sinusoides en el espacio perisinusoidal de Disse, aunque también pueden observarse entre las células parenquimatosas2-4. Clásicamente el espacio de Disse se define como la zona limitada por los hepatocitos y la pared sinusoidal formada por las células endoteliales sinusoidales4,5. Normalmente este espacio contiene fibras nerviosas y algunos componentes de la matriz extracelular (MEC) como, por ejemplo, fibras colágenas de tipos I y III y componentes de la membrana basal que, por lo general, no llegan a formar una membrana basal verdadera5-7.
En su estado quiescente las CEH constituyen el 5-8% del número total de células hepáticas considerando las parenquimatosas (hepatocitos), las endoteliales (tanto sinusoidales como vasculares), las células de Kupffer (macrófagos), las células epiteliales de la vía biliar y las CEH8-10. Su citoplasma se caracteriza por presentar múltiples y prominentes «gotitas» de grasa de 1 a 2 µm de diámetro2,3,11. Estas gotitas de grasa representan el principal sitio de almacenamiento de vitamina A (retinol). Más del 90% de la vitamina A hepática (el 80% del total del organismo) se halla almacenado en las CEH, mientras que el resto se encuentra en las células parenquimatosas12-14.
En las CEH, desde su pericarion nacen numerosas largas y finas prolongaciones citoplásmicas denominadas «procesos» o «prolongaciones primarias», que corren a lo largo de los sinusoides. De estas prolongaciones primarias surgen las prolongaciones secundarias, que entran en contacto directo con las células endoteliales15-17. Observaciones recientes revelan la presencia de «microespinas» que nacen de los procesos citoplásmicos y que interactúan con las células parenquimatosas delimitando un espacio entre éstas y las CEH donde se encuentran fibras colágenas y también fibras nerviosas. Estas observaciones pueden modificar el clásico concepto del espacio de Disse a otro definido como un espacio situado entre los hepatocitos, por un lado, y las CEH y el endotelio sinusoidal por el otro15.
En condiciones normales, las CEH cumplen varias funciones en el hígado como, por ejemplo, la ya mencionada función de almacenar la vitamina A, controlar la comunicación intercelular por medio de la liberación de varios mediadores intercelulares18-20 y remodelar gran parte de la poca MEC presente en un hígado normal por medio de: a) la producción de los componentes colágenos y no colágenos de la MEC20-22; b) la síntesis de las llamadas metaloproteinasas de la MEC (MMP, del inglés matrix metalloproteinase), que son un grupo de enzimas capaces de catabolizar los componentes de la MEC23,24, y c) la síntesis de los inhibidores de las MMP o inhibidores tisulares de metaloproteinasas (TIMP, del inglés tissue inhibitor of metalloproteinase), que tienden a controlar la actividad catalítica de las MMP a fin de mantener las sustancias de la matriz intersticial en un concentración estable25,26. Estas últimas funciones de las CEH tienen como propósito básico controlar la homeostasis tisular y mantener una especie de equilibrio fisiológico entre la síntesis y la degradación de los componentes de la MEC.
FISIOPATOGENIA DE LA FIBROGÉNESIS HEPÁTICA
La fibrogénesis hepática se define como un complejo y dinámico proceso fisiopatológico caracterizado por el depósito de tejido fibroso en el hígado como consecuencia de un desequilibrio entre la producción y la degradación de los componentes de la MEC en respuesta a varias agresiones repetitivas o crónicas de intensidades suficientes para conducir a este proceso de cicatrización. Independientemente de la causa, existe una progresiva deposición de múltiples tipos de proteínas colágenas y no colágenas en el intersticio y en el espacio de Disse.
Activación de las células estrelladas del hígado
Tras la lesión crónica del tejido hepático resultante de cualquier etiología, tanto las CEH como las otras células productoras de la MEC presentes en el tracto portal sufren un proceso anormal que se denomina «activación».
La activación de las CEH es un proceso patológico que se caracteriza por la pérdida de las típicas gotitas de grasa, el aumento del número y tamaño celulares y la transdiferenciación fenotípica a células proliferativas, fibrogénicas y contráctiles muy similares a los miofibroblastos. Tanto los estímulos patológicos como los mediadores intercelulares y los cambios de la MEC son factores de vital importancia para llevar a cabo dicha activación celular8,27,28.
El inicio del proceso de la activación de las CEH se produce por:
1. El impacto de la lesión de las células parenquimatosas o las células del tracto biliar sobre la homeostasis tisular.
2. Los efectos de los estímulos citocínicos provenientes tanto de células inflamatorias como de células vecinas.
3. Las modificaciones en la composición de la MEC, que las CEH perciben por medio de los receptores de aquélla, integrinas que conectan componentes de la MEC al citosqueleto27-30. Todas estas alteraciones provocan en las CEH importantes cambios en la expresión genética que generan una serie de modificaciones fenotípicas que les permiten responder a los crecientes estímulos patológicos locales. Estas modificaciones fenotípicas en las CEH activadas se sostienen por medio de una compleja y sofisticada señalización intercelular autocrina y paracrina19,31,32. Aunque la lista de los mediadores intercelulares que intervienen en este proceso se actualiza continuamente, la siguiente clasificación en grupos nos permite ordenar los mediadores intercelulares de mayor importancia.
Mediadores inductores de la proliferación y/o motilidad celulares: el factor de crecimiento derivado de las plaquetas, el factor de crecimiento básico de los fibroblastos y el factor de crecimiento insulinoide 1.
Mediadores fibrogénicos: los 2 más importantes son el factor transformador del crecimiento tipo b1 (TGF-b1) y la interleucina 6.
Mediadores inductores de la contracción de las CEH, tales como la endotelina-1, la trombina, la angiotensina II y la vasopresina.
Mediadores con actividad antiinflamatoria y antifibrogénica: la interleucina-10 y el interferón gamma.
También es importante recordar el papel del estrés oxidativo en la activación de las CEH. Varios estudios experimentales señalan que las especies reactivas de oxígeno, como el peróxido de hidrógeno (H2O2) y los radicales libres, están implicadas en el desarrollo y la progresión de diversas enfermedades como la esclerosis múltiple, el sida, la enfermedad de Alzheimer y la lesión hepática crónica, entre otros. Estos productos metabólicos son tóxicos para el organismo y conducen a alteraciones del metabolismo celular, inhibición del crecimiento y finalmente la muerte celular. Por fortuna el organismo dispone de un grupo de sistemas de enzimas antioxidantes para contrarrestar los efectos tóxicos de las especies reactivas de oxígeno24.
Según otros autores24,33, el proceso de activación de las CEH puede dividirse en 2 etapas, una de iniciación y otra de perpetuación. La iniciación (también llamada etapa preinflamatoria) corresponde a los primeros acontecimientos del proceso y comprende los cambios rápidos de la expresión genética y del fenotipo que permiten a las CEH responder a los estímulos locales y citocínicos; es el resultado de los estímulos paracrinos provenientes de las células vecinas y los cambios tempranos de la composición de la MEC.
La perpetuación corresponde a los acontecimientos celulares que amplifican el fenotipo activado por medio de los efectos crecientes de las diversas citocinas, y comprende la proliferación, fibrogénesis, contractilidad, realización de citocinas, quimiotaxis y motilidad celular, pérdida de los retinoides y regulación de la MEC. Esta etapa es el resultado de estímulos autocrinos y paracrinos, así como de la remodelación acelerada de la MEC.
REPERCUSIÓN DE LOS CAMBIOS FENOTÍPICOS DE LAS CÉLULAS ESTRELLADAS DEL HÍGADO ACTIVADAS EN LA FIBROSIS HEPÁTICA
Como ya se ha mencionado, la activación de las CEH conduce a varias modificaciones fenotípicas que lógicamente repercuten en el desarrollo de la fibrosis hepática.
Producción de la matriz intersticial
Normalmente la MEC del hígado consta de variadas sustancias colágenas y no colágenas cuyas composición y concentración son responsabilidad de las CEH. Estos componentes intercelulares son elementos esenciales para mantener la morfología y las funciones de las células del hígado y, como es obvio, sus alteraciones en términos cualitativos y/o cuantitativos determinan modificaciones patológicas tanto desde el punto de vista morfológico como funcional.
Como ya hemos adelantado, en la lesión hepatocelular las CEH sufren un proceso de transdiferenciación fenotípica a miofibroblastos. Estos «nuevos» miofibroblastos constituyen una «nueva» fuente de múltiples tipos de proteínas colágenas y no colágenas de la MEC, como el colágeno tipo I, tipo III, tipo IV, laminina, elastina, fibronectina y diversos proteoglucanos como la condroitinsulfato, el dermatansulfato y el heparansulfato. Por ejemplo, en las primeras etapas de la lesión hepatocelular ya existe una creciente deposición de colágeno tipo III, tipo IV y fibronectina en el espacio de Disse; no obstante, en el hígado cirrótico la cantidad de la MEC depositada por los miofibroblastos puede ser más de 6 veces la del hígado normal.
Muchas sustancias y agentes fibrogénicos provenientes de las células inflamatorias y de las células hepáticas, entre ellas las CEH, se liberan durante el proceso inflamatorio, donde actúan de forma paracrina y autocrina sobre las CEH y contribuyen a mediar y mantener el proceso fibrogénico. Uno de los más importantes y potentes mediadores profibrogénicos en la lesión hepática es el ya mencionado TGF-b34-36. Éste es un factor de crecimiento ampliamente distribuido en el organismo y regula no sólo la deposición de la MEC como parte de la respuesta normal a la lesión tisular, sino que también es un mediador de gran importancia en la fibrosis patológica37. El TGF-b pertenece a una familia de péptidos multifuncionales llamada superfamilia del TGF-b, que integra más de 30 citocinas, entre las cuales se incluyen las 5 isoformas del TGF-b, 3 de las cuales se identificaron en los mamíferos (TGF-b1, TGF-b2 y TGF-b3)38. Ejercen sus acciones a través de una familia de receptores membranosos tipos I y II de actividad proteincinasa intrínseca38,39. En el hígado estas 3 isoformas se encuentran presentes, aunque parece que el TGF-b1 es el de mayor importancia34,40 y tiene un papel muy destacado en la fibrogénesis hepática. El TGF-b es sintetizado principalmente por las CEH, donde produce de forma autocrina sus principales efectos fibrogénicos34. Los estudios in vitro e in vivo demuestran que el TGF-b provoca en las CEH activadas un aumento de la síntesis de las fibras colágenas, que se depositan progresivamente en el espacio intercelular y en el espacio de Disse, lo que altera la función y la homeostasis de las células hepáticas41,42. Asimismo estimula la síntesis de los componentes de la membrana basal, lo que conduce a la «capilarización» de los sinusoides (véase más adelante)43,44. En ratas transgénicas, la sobreexpresión del TGF-b1 puede inducir la fibrosis, mientras que la fibrosis experimental puede inhibirse con anticuerpos neutralizantes45,46. En la actualidad está ampliamente aceptado que la persistente estimulación autocrina de las CEH por el TGF-b es un mecanismo clave en el desarrollo de la fibrogénesis hepática34. Sin embargo, este concepto aún sigue en discusión.
Inhibición de la degradación de la matriz extracelular
La progresión de la fibrosis hepática se asocia no sólo a la excesiva producción de la MEC, sino además a una importante inhibición anormal de su degradación. Como hemos recordado antes, las MMP son un grupo de enzimas que se encargan de catalizar los componentes de la MEC. Los TIMP pueden bloquear las actividades catalíticas de estas enzimas a fin de mantener en condiciones normales un equilibrio fisiológico entre la síntesis y la degradación de los componentes de la MEC.
Dentro de las MMP cabe mencionar las colagenasas, que degradan los colágenos intersticiales (tipos I, II y III); las gelatinasas, que se encargan de catalizar componentes de la membrana basal, y las estromalisinas, que hacen lo mismo sobre un amplia serie de sustancias como los proteoglucanos, la laminina, etc.47-49. Todas esas MMP son sintetizadas por diferentes tipos de células hepáticas50, mientras que el TIMP-1 y el TIMP-2, que son los 2 inhibidores de las MMP más importantes y estudiados hasta el momento, se expresan únicamente en la CEH51,52. Estas moléculas en conjunto son de mucha importancia no sólo en la fisiología de la remodelación y reparación de la MEC, sino también en el desarrollo del proceso fibrogénico.
En cultivo primario de CEH, la expresión de MMP como las colagenasas y las estromalisinas aumenta transitoriamente en la primera etapa del cultivo, para luego disminuir hasta volverse indetectable, mientras que tanto la expresión del TIMP-1 como la del TIMP-2 aumentan llamativa y progresivamente en dicho cultivo53-55. En modelos experimentales de animales con fibrosis hepática inducida por medio de la ligación del conducto biliar o el tetracloruro de carbono (CCl4), la expresión del TIMP-1 y del TIMP-2 aumenta significativamente entre las 3 y 6 h después de haberse producido la lesión hepatocelular, para luego permanecer en aumento progresivo a lo largo de la fase crónica de dichos modelos52,55.
En pacientes con enfermedades hepáticas crónicas como la colangitis esclerosante, la cirrosis biliar primaria, la hepatitis crónica autoinmunitaria activa y la atresia biliar existe un importante aumento de las concentraciones tisulares de TIMP-1, TIMP-2 o ambos; dichos valores pueden ser 5 veces más elevados que en el hígado normal54,56, lo que indicaría que la expresión de los TIMP por las CEH favorece aún más el depósito del tejido fibroso.
Capilarización de los sinusoides
Durante el proceso fibrogénico, el sistema microvascular hepático se modifica: de un sistema sinusoidal de capilares discontinuos, pasa a ser un sistema de capilares continuos; es lo que se denomina «capilarización»57-60. Hay un aumento de la producción de colágeno tipo IV, laminina y entactina que se depositan en el espacio de Disse, donde forman una membrana basal verdadera61,62. Las observaciones al microscopio electrónico revelan una importante disminución de las fenestraciones endoteliales tanto en número como en tamaño (defenestración) y la formación clara de una membrana basal verdadera de ubicación subendotelial63. Algunos autores incluyen, como una etapa siguiente a la capilarización, la «venulización», que se define como la transformación de los sinusoides en vénulas mediante el aumento de su diámetro, la pérdida de las fenestraciones y la formación de una membrana basal subyacente34. Como normalmente muchas funciones hepáticas dependen del intercambio rápido y bidireccional de macromoléculas entre el plasma y los hepatocitos, la capilarización altera la difusión y la filtración de sustancias bioactivas entre estos 2 compartimentos.
Contracción de las células estrelladas del hígado y regulación del flujo sanguíneo sinusoidal
Las CEH presentan características morfológicas, ultraestructurales y funcionales similares a los pericitos, que participan en la regulación del flujo sanguíneo microvascular en diversos órganos65-67. Las CEH adoptan una estratégica ubicación perisinusoidal; extienden a partir de sus cuerpos celulares varias prolongaciones primarias de 20 µm de longitud que corren a lo largo de uno o más sinusoides. De estas prolongaciones primarias surgen perpendicularmente prolongaciones secundarias, que tienden a rodear los sinusoides de forma circular, lo que apuntaría a un posible e interesante papel de las CEH en el control del flujo sanguíneo15,17. Estudios con microscopia intravital de hígados aislados de rata revelan que las CEH pueden mediar el diámetro sinusoidal en presencia de mediadores vasoactivos68. Además, se ha demostrado in vitro que la contracción y relajación de las CEH se producen efectivamente en respuesta a diversos mediadores vasoactivos como las endotelinas, la angiotensina II, el óxido nítrico (NO) y el péptido natriurético arterial, entre otros69-72.
Pero, más allá de estos datos, aún no existen claras evidencias de la participación de las CEH en el control de la perfusión del hígado en condiciones fisiológicas. Sin embargo, cuando hay fibrosis hepática patológica, parece que las CEH influyen activamente en el manejo del flujo sanguíneo sinusoidal y, por lo tanto, en la fisiopatología de la hipertensión portal. La aparición de estructuras contráctiles como los cuerpos densos y la actina alfa del músculo liso73,74, las modificaciones del microambiente por los cambios de la MEC, que conduce a una mayor tensión citosquelética75,76, y la aparición de una importante expresión de canales de calcio dependientes de voltaje en las CEH activadas, que favorecen un importante incremento de la concentración de Ca2+ intracelular, seguido por la contracción celular77,78, son algunas de las evidencias ultraestructurales que sostienen lo anterior. Estas modificaciones se observan sólo en las CEH activadas, no en las quiescentes.
Principales estimulantes de la contracción de las células estrelladas del hígado
Endotelinas. Son de los vasoconstrictores más potentes conocidos hasta ahora. Se han identificado 3 tipos de esa familia (endotelinas 1, 2 y 3), que actúan sobre 2 tipos de receptores: los receptores tipo A y los receptores tipo B79,80. De estas 3 isoformas, la endotelina-1 es la más estudiada en el hígado. Se sintetiza mayoritariamente por las CEH y actúa en diversas células hepáticas, aunque los receptores de las endotelinas abundan claramente en las CEH activadas81,82. A través de los receptores A, la endotelina-1 produce en las CEH un aumento transitorio de la concentración de Ca2+ intracelular (más de 8 veces por encima de los valores basales) unido a un reversible proceso de contracción celular. In vitro se produce la contracción del 78, el 55, el 59 y el 56% de las CEH después de exponerlas a 10 nM de endotelina-1 durante 2, 5, 10 y 20 min, respectivamente, en contraste con la relajación del 82, el 83 y el 71% de las CEH contraídas después de añadir 500 µM de nitroprusiato de sodio72.
La estimulación de estas células con endotelina-1 produce su contracción dentro de 10 s y alcanza una fuerza máxima de contracción en 5 min, mientras que al retirar esa sustancia del medio la relajación completa o casi completa de las CEH se da dentro de 45 min17. También la endotelina-1 disminuye el diámetro sinusoidal en un 25% y aumenta el gradiente de presión sinusoidal en un 116% y la resistencia vascular en un 350% en hígados aislados de ratas observados con microscopia intravital68. En el modelo de hígado de rata aislado y perfundido, la infusión de la endotelina-1 provoca, de forma dependiente de la dosis, un aumento de la presión portal asociado a un notable incremento de la glucogenólisis y del consumo de oxígeno83. Es importante tener en cuenta que la contractilidad de las CEH inducida por la endotelina-1 aumenta progresivamente a medida que avanza la lesión hepática y es directamente proporcional al grado de activación de las CEH, siendo más prominente en la cirrosis84. Los antagonistas de los receptores A y B de la endotelina pueden disminuir la presión portal en animales con hipertensión portal por un efecto inhibitorio sobre la contracción de las CEH85.
Angiotensina II. Desempeña un papel importante en la vasoconstricción por la interacción con el receptor de la angiotensina I86.
En las CEH activadas existe una alta expresión de los componentes del sistema renina-angiotensina, tales como la renina activa, la enzima de conversión de la angiotensina y la angiotensina II87-90. La interacción de la angiotensina II con la angiotensina I provoca varias acciones y efectos de mucha importancia fisiopatológica; de ellos, uno de los más importantes es el incremento, dependiente de la dosis, de la concentración del Ca2+ intracelular, con estimulación de la contracción celular, lo que produce hipertensión portal. Los antagonistas de la angiotensina II como el losartán bloquean estas acciones y mejoran la hipertensión portal91-93. Todas estas observaciones apenas pueden detectarse en las CEH quiescentes.
Hormona antidiurética. Es un potente vasopresor producido por la neurohipófisis y actúa sobre receptores específicos V1, V2 y V394. En el hígado produce diversos efectos sobre los receptores V1, recientemente identificados en las CEH95. Estos efectos son muy similares a los observados sobre las células del músculo liso vascular, tales como el aumento de la concentración del Ca2+ intracelular y el estímulo de la contracción celular de forma dependiente de la dosis. Estos efectos pueden bloquearse por medio de antagonistas del receptor V1.
Principales relajantes de la contracción de las células estrelladas del hígado
Fisiológicamente, la contracción de las CEH queda contrarrestada por los efectos de agentes vasorrelajantes como el NO, el péptido natriurético arterial, entre otras sustancias que se detallan en la tabla I. De estos mediadores, el más estudiado es el NO, que participa en diversos procesos como la regulación del tono vascular, la neurotransmisión y la citotoxicidad mediada por células96-98. Se sintetiza por la conversión de la L-arginina a L-citrulina por medio de la NO sintetasa99,100.

En el hígado, estudios in vitro e in vivo revelan que el NO es sintetizado en el endotelio por la NO sintetasa constitutiva. Se ha observado un aumento de la presión portal en animales que reciben de forma intraperitoneal un inhibidor específico de la biosíntesis del NO (el N-w-nitro-L-arginina), y por ello se ha relacionado el NO con la regulación de la circulación portal intrahepática en el lecho vascular del hígado normal de ratas101. La microcirculación en el hígado cirrótico se caracteriza por la disfunción endotelial provocada por la menor liberación de factores endoteliales relajantes, entre ellos el NO, lo que produce una menor vasodilatación y un significativo aumento de la resistencia intrahepática en pacientes con hipertensión portal102. Otros estudios demuestran que la lesión hepatocelular conduce a un aumento de la producción de la endotelina-1 y a una disminución de la síntesis del NO; este fenómeno favorece la constricción de los sinusoides y las paredes vasculares por la actividad contráctil de las CEH y las células musculares lisas, respectivamente, lo que contribuye a un incremento en la resistencia intrahepática y de la presión portal102.
NOCIONES GENERALES SOBRE LAS FUTURAS ESTRATEGIAS TERAPÉUTICAS
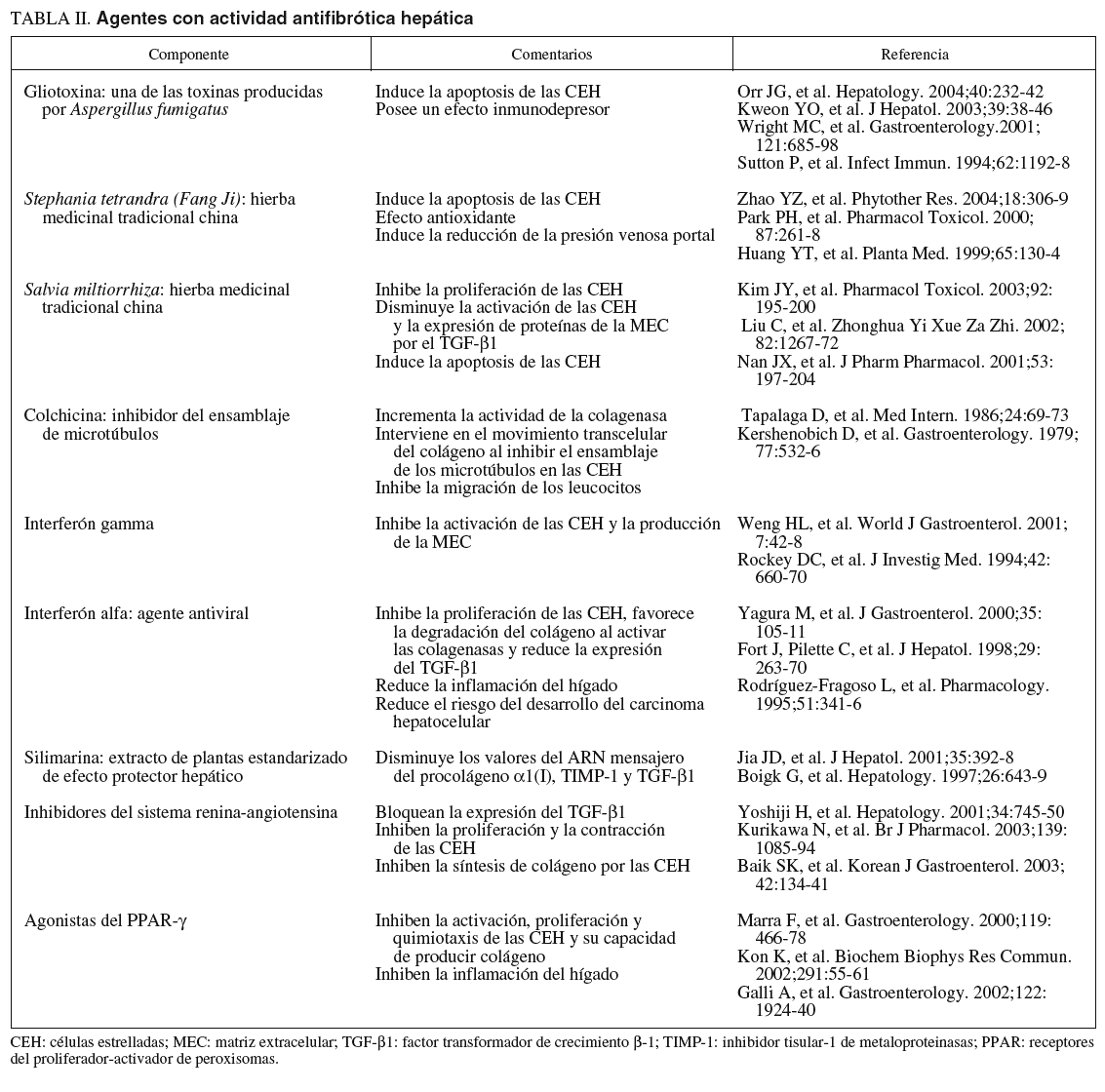
Las CEH han sido un atractivo e interesante blanco para el desarrollo de agentes antifibróticos que puedan resolver el problema de la fibrosis y cirrosis hepáticas. Muchos científicos han planificado interesantes líneas de investigación como, por ejemplo, reducir el proceso inflamatorio y la respuesta inmunitaria, inhibir la activación de las CEH, inducir la apoptosis de las CEH activadas, bloquear la actividad fibrogénica, contráctil, proliferativa y proinflamatoria de las CEH, disminuir la síntesis de los componentes de la MEC y/o favorecer su degradación103. En la tabla II se mencionan algunos de los más importantes componentes «antifibrogénicos» en estudio.
La carencia de marcadores séricos establecidos para evaluar la respuesta hepática a los fármacos en estudio obliga al investigador a realizar varias biopsias hepáticas a los pacientes cirróticos en tratamiento, lo que es poco agradable para el paciente104. La falta de selectividad celular in vivo de los fármacos que muestran un efecto antifibrótico in vitro es otro importante problema de los medicamentos en uso, que pueden terminar produciendo importantes efectos secundarios, como la apoptosis de los macrófagos y linfocitos T por el uso de la gliotoxina105, el riesgo de desarrollar enfermedades autoinmunitarias y neoplásicas por el empleo de antagonistas del TGF-b106,107, el riesgo de desarrollar tumores malignos por el uso de los agonistas de los receptores del proliferador-activador de peroxisomas108,109, etc. Surge entonces la necesidad de crear fármacos antifibróticos «inteligentes» que puedan actuar selectivamente en las CEH activadas.

