




UN RECHAZO, PERO AL REVÉS
El trasplante alogénico de precursores hemopoyéticos (TPH) es un proceso terapéutico ampliamente utilizado en los últimos 30 años, empleado como tratamiento de rescate en pacientes afectados de leucemias agudas y crónicas (fundamentalmente mieloides) y de otras neoplasias metastásicas, después de tratamientos antitumorales letales o subletales. También se ha utilizado como tratamiento de relleno o sustitución en aplasias medulares graves o situaciones dishemopoyéticas graves no tumorales (p. ej., en la talasemia mayor)1-3.
El trasplante de órganos sólidos se puede complicar con una reacción del huésped contra el injerto cuando las células inmunocompetentes del receptor reconocen como extraños alguno(s) de los antígenos de histocompatibilidad del injerto y lo rechazan y destruyen. En los TPH esta reacción de disparidad antigénica se invierte, ya que el receptor, antes de recibir la suspensión de precursores hemopoyéticos, se ha sometido a un curso corto e intenso de radioterapia, en forma de irradiación corporal total, o de quimioterapia muy agresiva, que reciben la denominación de «tratamiento de acondicionamiento». El fin de esta última es provocar al mismo tiempo, en el huésped, la destrucción de las células no deseadas y la depresión inmunológica que impida el rechazo del injerto.
De esta manera, cuando existe disparidad entre los antígenos de histocompatibilidad del donante y el huésped, se puede originar una reacción entre los linfocitos T (LT) maduros que acompañan al inóculo de precursores hemopoyéticos del donante y ciertos tejidos del huésped. Esta reacción, o rechazo al revés, es la que se conoce como enfermedad del injerto contra el huésped (EICH)3-6. La disparidad antigénica que provoca esta reacción no sólo se refiere a antígenos del complejo principal de histocompatibilidad (CPH) o sistema HLA de los humanos, sino también a los llamados antígenos menores de histocompatibilidad, todavía mal conocidos3-7,8. La EICH es más agresiva cuando el tratamiento de acondicionamiento es mieloablativo.
La EICH se presenta bajo 2 formas clinicobiológicas diferentes9-11. La forma aguda, que surge durante los primeros 2-3 meses tras el trasplante, afecta fundamentalmente a 3 territorios de la economía: la piel, el sistema hepatobiliar y el tracto intestinal. Por otra parte, la forma crónica aparece después de aquel período de tiempo como un proceso multiorgánico con características de enfermedad autoinmunitaria.
SECUENCIA MULTIFÁSICA DE LA ENFERMEDAD DEL INJERTO CONTRA EL HUÉSPED AGUDA INTESTINAL
Por razones biológicas, hoy por hoy desconocidas, las manifestaciones clínicas de la EICH aguda quedan secuestradas muy preferentemente en los 3 territorios antes comentados.
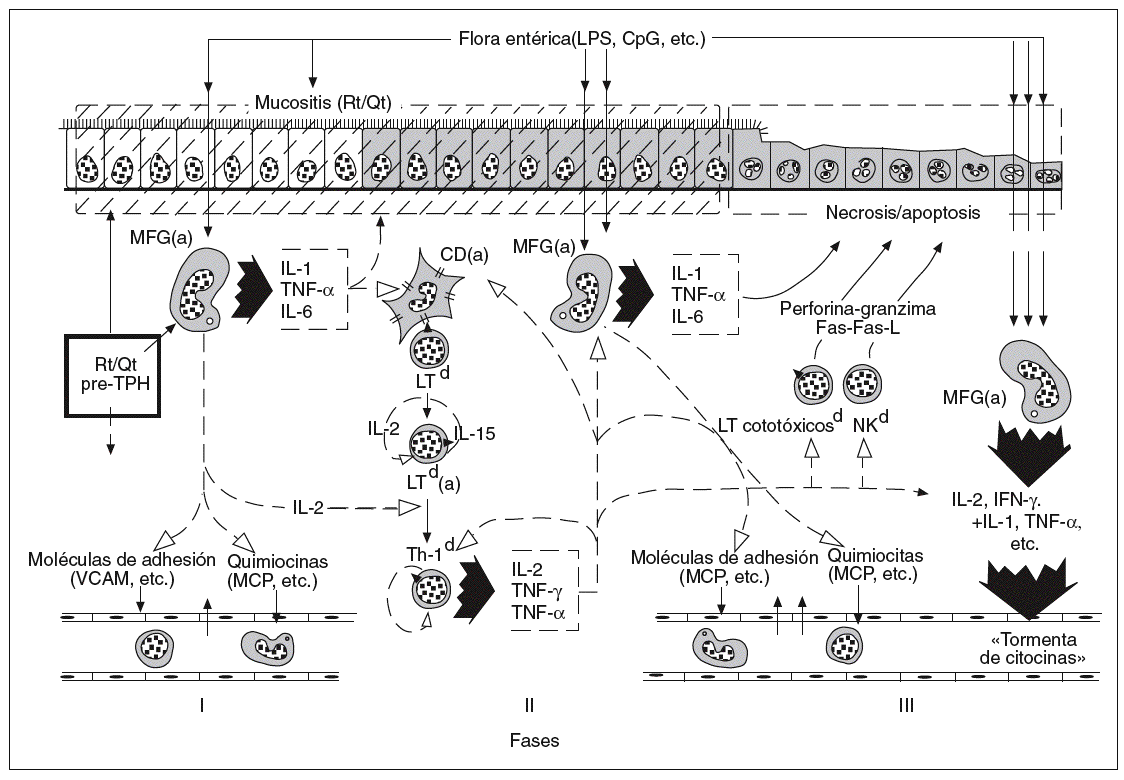
En los últimos años ha empezado a comprenderse mejor, tanto en clínica humana como en patología experimental, la importancia del intestino en la secuencia de acontecimientos fisiopatológicos que explican la patogenia de la EICH aguda12-14. Con fines de sistematización didáctica, se ha fraccionado aquella secuencia en 3 fases15: la de inducción pretrasplante, la de activación de LT del donante y, finalmente, la fase efectora de citotoxicidad y la fase final de la «tormenta de citocinas»9, que se inicia con la activación y expansión clonal de LT del donante y alcanza su máxima expresión y diseminación al final de la secuencia patogénica (fig. 1). A cada una de estas fases hemos dedicado los apartados siguientes de este trabajo, con especial referencia al protagonismo intestinal en cada una de ellas.
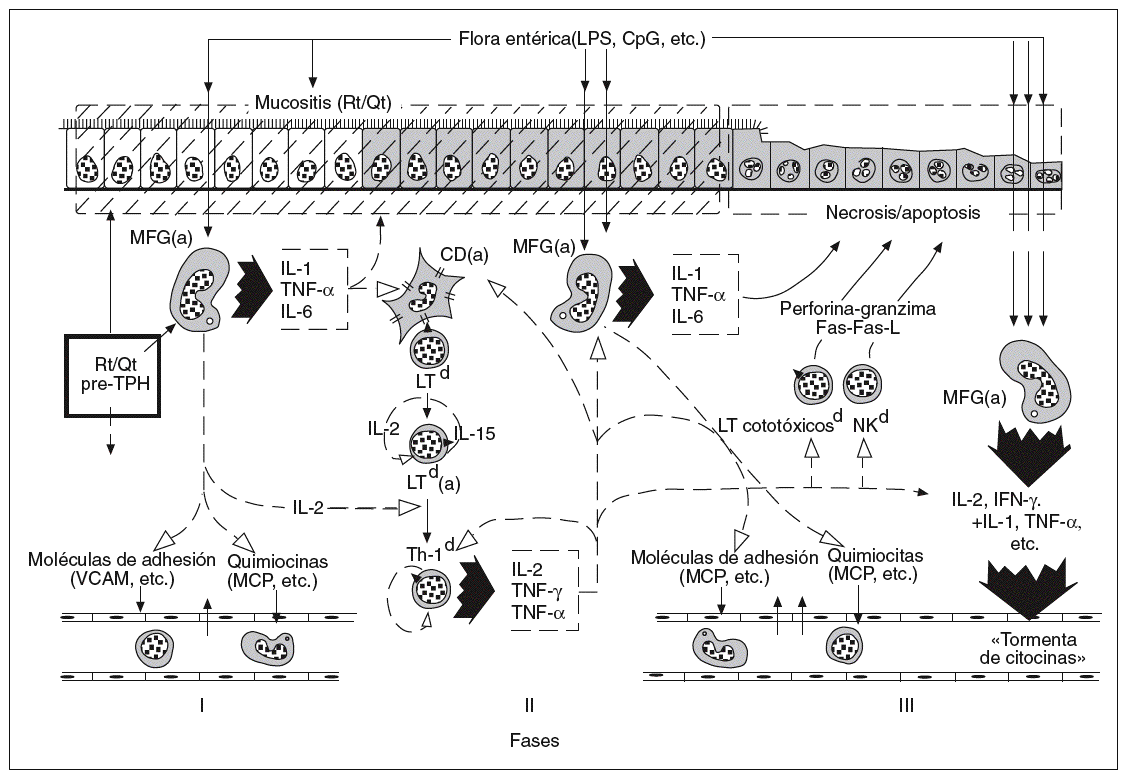
Fig. 1. Secuencia multifásica de la enfermedad del injerto contra el huésped aguda intestinal. CpG: citosina-fosfodiesterasa-guanina; Rt: radioterapia; Qt: quimioterapia; TPH: trasplante alogénico de precursores hemopoyéticos; MFG: macrófagos; IL: interleucina; TNF: factor de necrosis tumoral; VCAM: molécula de adhesión vascular; MCP: proteína quimitáctica de monocitos; LT: linfocitos T; Th: linfocitos T helper; NO: óxido nítrico; IFN; interferón; NK: células citolíticas (natural killer).
FASE I: INDUCCIÓN PRETRASPLANTE
Los tejidos del paciente que en breve va a recibir el TPH se encuentran sometidos al estímulo de diversos factores, como son: a) la enfermedad de fondo (leucemia, neoplasia metastásica, etc.); b) los tratamientos previos de ésta; c) las complicaciones infecciosas, y sobre todo d) el tratamiento de acondicionamiento que precede al trasplante (radiación corporal total o quimioterapia agresiva)14,16,17.
Entre las células más sensibles a esta activación pretrasplante se encuentran los elementos del sistema inmunitario innato18-20, fundamentalmente los macrófagos y las células dendríticas tisulares, y entre éstos, los que habitan en la lámina propia de la mucosa intestinal (fig. 1), formando parte del tejido linfoide terciario asociado a mucosas.
Por otra parte, el tratamiento de acondicionamiento lesiona el epitelio de la mucosa intestinal. Esta mucositis yatrógena provoca un aumento de la permeabilidad de la barrera de enterocitos al paso de productos bacterianos de origen intraluminal a los espacios subepiteliales de aquélla (donde activan, maduran y hacen proliferar a los macrófagos y células dendríticas) y también a la circulación sanguínea general8,13.
Entre estos productos se encuentran los lipopolisacáridos de las bacterias gramnegativas normales del intestino, que se comportan como endotoxinas y que recogen receptores específicos en aquellas células. Unos receptores, localizados en la membrana, pertenecen a la familia Toll-like (TLR-4), y otros son receptores citosólicos, pertenecientes a la familia NOD (rNOD-2)19.
Otro producto bacteriano, con amplia capacidad inmunoestimuladora sobre macrófagos y células dendríticas, es el oligonucleótido no metilado citosina-fosfodiesterasa-guanina (CpG), captado gracias a otro receptor de membrana de aquellas células, miembro también de la familia Toll-like (el TLR-9)19,21.
Las señales biológicas incorporadas por los receptores membranarios (TLR-4 y TLR-9) y citosólicos (rNOD-2) se transmiten en forma de mensajes intracelulares convergentes. Todos ellos parecen conducir a la activación del llamado factor nuclear kappa-beta, que actúa sobre algunos genes encargados, en aquellas células, de la síntesis de citocinas y otras moléculas promotoras de la inflamación (local y a distancia), como son: el factor de necrosis tumoral alfa (TNF-a), varias interleucinas (IL-1, IL-6, IL-12, etc.) y el factor estimulante de colonias granulomonocíticas, entre otras8,16,22.
La liberación de esta primera oleada de citocinas (sobre todo TNF-a) origina una serie de efectos, como son8,17:
a) el incremento de la expresión de moléculas de adhesión vascular (VCAM), plaquetaria (PECAM), etc. y de algunas quimiocinas leucoatrayentes (proteína quimiotáctica de monocitos, etc.), lo que facilita la llegada a los espacios subepiteliales de la pared intestinal de leucocitos para participar en la «batalla inflamatoria» que empieza a librarse; b) la hiperexpresión membranaria de los antígenos del CPH, tanto en las células presentadoras de antígenos «profesionales» (células dendríticas), «semiprofesionales» (macrófagos) o «eventuales» (como son los propios enterocitos), y finalmente c) aquellas citocinas (TNF-a e IL-1) tienen capacidad directa para lesionar la barrera epitelial, hecho que agrava la mucositis originada por la radioterapia/quimioterapia del tratamiento de acondicionamiento.
De esta manera, se hace más permeable la frontera mucosa a la translocación de productos bacterianos, desde la luz intestinal (lipopolisacáridos, CpG, etc.), y con ello comienza a cerrarse el «bucle de acontecimientos» que desde el intestino actúa como inductor pretrasplante de la EICH aguda en general13,16,17.
FASE II: ACTIVACIÓN DE LINFOCITOS T
DEL DONANTE
El incremento de la expresión de los antígenos del CPH por parte de las células presentadoras de antígenos del receptor (células dendríticas, macrófagos y enterocitos), originado al final de la fase anterior8,17, hace más fácil el reconocimiento de la disparidad antigénica por parte de los LT del donante. En este contexto, tiene lugar el
segundo episodio del «arco aferente» de la EICH aguda intestinal y momento crucial de ésta, que, en síntesis, significa la activación (seguida de proliferación y diferenciación citofuncional) de aquellos linfocitos como alorreacción frente a los antígenos extraños que aparecen frente a ellos: las células del receptor8,15-17,23.
Es obvio que después del TPH la primera frontera de contacto y enfrentamiento entre aloantígenos del receptor y los LT del donante debe ser el propio endotelio capilar. Por esta razón, se está investigando el posible papel de antígenos menores de histocompatibilidad de las moléculas de adhesión vascular hiperexpresadas en el receptor durante la fase I de la EICH aguda8. Entre éstas se ha prestado especial atención a la llamada PECAM-1 (CD31), cuyo gen codificador pertenece a la superfamilia de genes de las inmunoglobulinas. Todo ello apunta a que diferencias donante/receptor en estas moléculas pueden ser importantes en la patogenia de la EICH aguda, aunque los datos disponibles son todavía escasos.
En líneas generales, se puede decir que, frente a disparidades entre antígenos de clase I del CPH (antígenos HLA-A, B y C), son los LT-CD8+ del donante los que
reaccionan, de manera directa y contundente, como linfocitos citotóxicos, tal como veremos más adelante. Por el contrario, frente a disparidades entre antígenos de clase II (DR, DP y DQ) y sus péptidos asociados responden preferentemente los LT-CD4+ del donante8,15-17,24.
La oferta antigénica mejor conocida, tanto en patología humana como experimental, es la que realizan las células presentadoras de antígenos (fundamentalmente las células dendríticas) del huésped, a través de las moléculas antigénicas del CPH de clase II, a los LT-CD4+ del donante. La primera señal de esta activación la pone en marcha el acoplamiento: a) por parte de las células dendríticas, del complejo formado por los antígenos (DR, DP y DQ) y los péptidos antigénicos que llevan engarzados (que se comportan como antígenos menores de histocompatibilidad), y b) del receptor membranario de los LT-CD4+ (rCT), genéticamente preparado para reaccionar frente a aquel complejo molecular8,15,17,25.
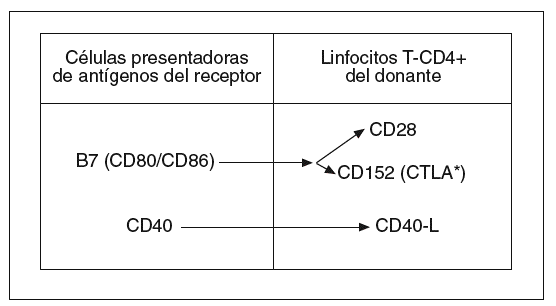
Sin embargo, parece que es necesaria una segunda señal, que procede de moléculas coestimuladoras situadas en la membrana de las células presentadoras de antígenos del receptor y de los LT-CD4+ del donante (fig. 2). Parece ser que, cuando la coestimulación iniciada por la molécula B7 de las células presentadoras de antígenos se canaliza a través de la molécula linfocitaria CD28, se liberan señales positivas que favorecen la activación linfocitaria. Lo contrario ocurre cuando aquella señal se canaliza a través de la molécula CTLA (antígeno linfocitario citotóxico)8,23.
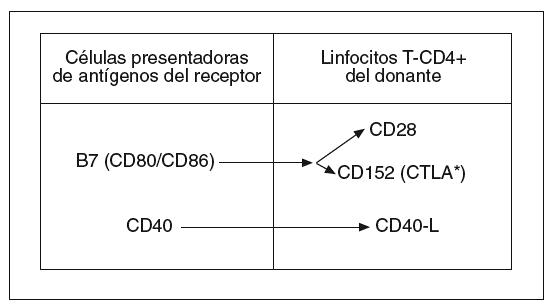
Fig. 2. Moléculas coestimuladoras causantes de la segunda señal de activación de linfocitos T en la enfermedad aguda del injerto contra el huésped. CTLA: antígeno linfocitario citotóxico.
Esta activación pone en marcha eventos moleculares como la secreción, por parte de los LT, de citocinas como la IL-2 e IL-15 y la expresión membranaria de sus respectivos receptores. Así, por un camino de estimulación autocrina, se induce la expansión clonal de los LT alorreactivos15,17,23.
Después de esta fase de proliferación postactivación, algunos LT alorreactivos pueden eliminarse a través de un proceso de deleción, quizá como un intento por amortiguar la gravedad de la reacción del injerto contra el huésped que se avecina. Parece tratarse de un mecanismo delecional periférico de muerte celular inducida por activación. Esta apoptosis periférica se ve facilitada por la molécula membranaria Fas (hiperexpresada en los LT activados) cuando estos linfocitos entran en contacto con células que expresan en su membrana el ligando de aquella molécula (Fas-L)16,17,26,27.
Hoy sabemos que, en el último escalón de esta fase II de la EICH, los LT-CD4+ activados tienen opción a 2 caminos de diferenciación citofuncional: bien hacia una sublínea implicada en las respuestas inmunitarias de «tipo celular» la de los LT helper (Th-1), con la consiguiente activación transcripcional de los genes que codifican la síntesis de citocinas proinflamatorias, como el interferón gamma (IFN-g), la IL-2 y el TNF-a, bien hacia la sublínea Th-2, que colabora y facilita las respuestas inmunitarias de tipo humoral, secretora fundamentalmente de IL-4, IL-5 e IL-1014,15,17,28.
Por estas razones biológicas desconocidas, parece que en la EICH aguda la diferenciación preferente de los LT alorreactivos se realiza en sentido Th-1, hecho facilitado por la IL-12 segregada por los macrófagos en la fase anterior. Por lo tanto, se liberan sobre todo citocinas proinflamatorias, como la IL-2, el IFN-g y el TNF-a15-17,23.
Estas citocinas Th-1 retroalimentan la fase II de la EICH aguda en sus órganos diana, entre ellos el intestino, y lo hacen: a) autoestimulando la capacidad proliferativa de esta sublínea Th-1; b) activando las células presentadoras de antígenos del receptor (células dendríticas y macrófagos), en las que incrementan su sensibilidad frente a los productos bacterianos de origen intestinal (lipopolisacáridos y CpG); c) aumentando la expresión membranaria de los antígenos del CPH del receptor, y d) incrementando la lesión de mucositis intestinal mediante la acción del IFN-g, que estimula la secreción de la IL-1 y del TNF-a, por parte de los macrófagos, con capacidad citotóxica, con lo que el flujo de aquellos productos bacterianos de origen intraluminal continuará progresando16,17.
FASE III: CITOTOXICIDAD Y TORMENTA FINAL
Ésta es la fase final de la EICH aguda, en que se lesionan las células diana de esta reacción precoz de los LT8,15-17. Se trata de una fase muy compleja en la que cooperan sinérgicamente toda una serie de citocinas inflamatorias y otras moléculas solubles que se han ido generando a lo largo del proceso, junto con ciertos efectores celulares con actividad citotóxica. Para no perdernos, los comentaremos por separado, a sabiendas de que actúan en íntima conexión.
La activación alorreactiva de los LT, con diferenciación citofuncional en sentido Th-1, termina con la secreción de ciertas citocinas proinflamatorias (IL-2, IFN-g, etc.) que no sólo retroalimentan esta fase II de la EICH aguda intestinal, sino que siguen estimulando la actividad secretora de los macrófagos locales (IL-1, TNF-a, óxido nítrico, etc.), lo que incrementa la exposición, en el endotelio capilar, de moléculas de adhesión vascular y quimiocinas leucoatrayentes.
Algunas de aquellas moléculas, como el TNF-a, pueden lesionar el epitelio intestinal, como hemos visto antes, provocando citonecrosis y apoptosis15-17,29. La IL-1 desempeña un papel sinérgico con el TNF-a en la EICH aguda16,17,30. A su vez, el óxido nítrico es otro factor inflamatorio de origen macrofágico que, al producir la liberación de hierro de las células diana intestinales, impide la recuperación del epitelio lesionado al inhibir la actividad proliferativa de los progenitores criptales del intestino16,17,31.
Parece también que el IFN-g induce la hiperexpresión membranaria de los antígenos del CPH en los propios enterocitos; de esta manera hace más clara la diana final a la acción citotóxica de los efectores celulares de la EICH aguda intestinal.
Finalmente, las citocinas proinflamatorias segregadas por los LT-CD4+ en su diferenciación Th-1 (IL-2 e IFN-g) estimulan la actividad de los principales efectores celulares de la reacción que nos ocupa: concretamente, los LT citotóxicos y las células citolíticas del donante8,15.
Estas células efectoras de la fase III de la secuencia patogénica de la EICH aguda intestinal parecen actuar sobre el epitelio diana a través de: a) la vía molecular clásica de citólisis, es decir, la de la perforina-granzima, y b) la vía apoptótica mediada por el acoplamiento de las moléculas Fas-Fas-L8,16,17,32-34.
Parece que los LT citotóxicos CD8+ (y también las células citolíticas) utilizan preferentemente el camino protagonizado por la perforina-granzima, que son moléculas almacenadas en los gránulos citoplásmicos de estos LT y también de las células citolíticas8,17. Tras la aproximación y el reconocimiento de las células diana (a través de la interacción del CPH y el rCT) la perforina segregada por aquellas células se inserta en la membrana de los enterocitos diana y la perfora, con lo que se producen los llamados «poros de perforina». Estos poros permiten la entrada en dichos enterocitos de las granzimas A y B, y con ello la citólisis, previa activación de las caspasas. Por el contrario, parece ser que los LT citotóxicos CD4+ utilizarían preferentemente la vía patogénica generada por el acoplamiento de las moléculas Fas-Fas-L, hecho que conduce a la formación de un complejo inductor de la muerte celular o apoptosis8,17.
Así, a través de estas 2 vías moleculares se produce, fundamentalmente, la citotoxicidad del epitelio intestinal diana (necrosis/apoptosis), desde la actividad agresiva de los LT citotóxicos y de las células citolíticas del donante, que trabajan sinérgicamente con las moléculas inflamatorias solubles (TNF-a, etc.) procedentes del sistema mononuclear fagocítico del receptor y quizá también del propio trasplante16.
Los fenómenos de necrosis y apoptosis de las células epiteliales del intestino, como expresión de la fase III de la secuencia patogénica de la EICH aguda en el aparato digestivo, terminan por abrir todavía más la barrera de los enterocitos, progresivamente desvencijada por los acontecimientos de las fases I y II que sobre ella actúan. De esta manera se permite el paso en cascada de los productos bacterianos de origen intraluminal, que terminan activando al máximo los elementos del sistema mononuclear fagocítico (tanto del receptor como del donante) con la producción masiva de citocinas inflamatorias que, sumadas a las que fabrican los LT del donante, activados en la fase II, terminan por cuajar una auténtica «tormenta de citocinas» que amplifica la EICH aguda sistémica8,15.
REFLEXIONES FINALES
Es bien conocido que la pared intestinal es el mayor órgano linfoide de nuestra economía, ya que acumula, en forma de agregados linfoides o linfocitos sueltos, un 25% de todos los inmunocitos humanos14. Si bien cuantitativamente esto es muy importante, aún lo es más el hecho cualitativo de que esta población celular se asocia estrechamente a un epitelio muy especializado, cuyo correcto funcionalismo es imprescindible para la supervivencia de nuestra especie.
Por todo lo anteriormente expuesto, no es de extrañar que cualquier reacción inmunológica que tenga como diana al intestino, como ocurre en la EICH aguda, pueda beneficiarse a veces de su papel «inductor» y con frecuencia de su papel «amplificador».
Este protagonismo del intestino en la EICH, sustentado fundamentalmente por investigaciones en patología experimental, ha dado pie al intento de proporcionar un «blindaje molecular» del epitelio intestinal que lo protegiese de los efectos lesivos del tratamiento de acondicionamiento pretrasplante, de la translocación de productos bacterianos de origen intestinal y de los acontecimientos biológicos que terminan con la «tormenta de citocinas»12,13,23,35. Entre estas moléculas protectoras destacan la IL-11, el factor de crecimiento queratinocítico y quizá también la IL-7.
Si los efectos proliferativos sobre la EICH aguda murina logrados con estos agentes pudiesen extrapolarse a la patología humana, es evidente que su violenta expresión clínica podría beneficiarse del «enfriamiento» de la pared intestinal en su papel «inductor» y «amplificador» de aquélla.

