




La leiomiomatosis peritoneal diseminada es una enfermedad benigna e infrecuente, de la que existen unos 150 casos descritos en la literatura1. Se caracteriza por múltiples nódulos constituidos por músculo liso en la cavidad abdominopélvica. Normalmente aparece en mujeres premenopáusicas2,3, y en algunos casos se ha descrito relación con estados de hiperactividad hormonal como la toma de anticonceptivos orales, neoplasia de ovario, etc.1,4,5. En la mayoría de casos son asintomáticas, aunque la clínica dependerá de número y tamaño de las lesiones, así como un crecimiento rápido. Su diagnóstico suele ser un hallazgo casual en un estudio radiológico o en el transcurso de una intervención quirúrgica1,5. El mayor problema es establecer el diagnóstico diferencial con la carcinomatosis peritoneal3,5, siendo su diagnóstico definitivo histológico2.
El tratamiento debe individualizarse, dependiendo de las características y la sintomatología de la paciente.
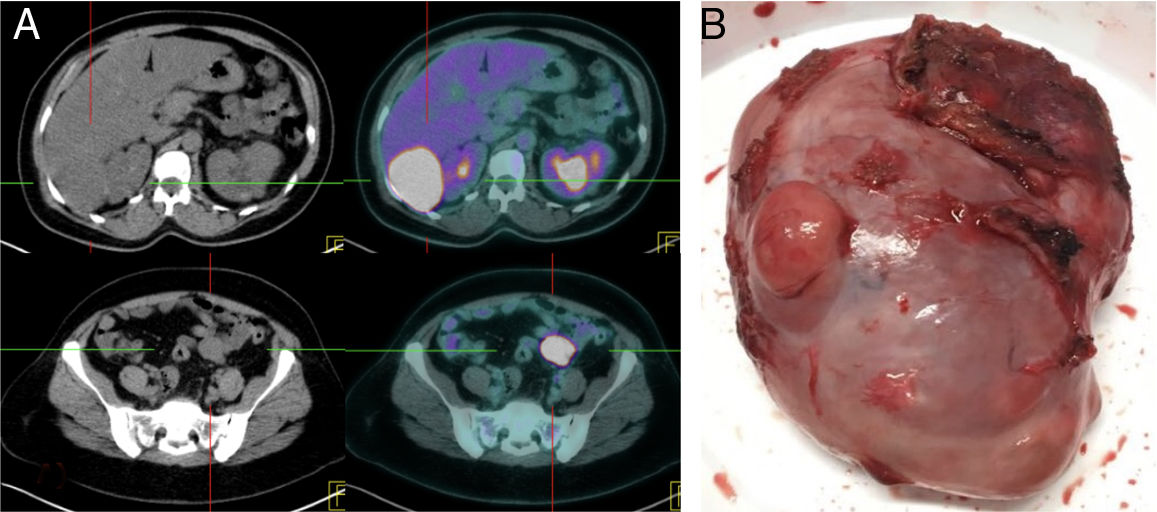
Presentamos el caso de una mujer de 42 años con antecedentes de HTA, poliquistosis renal en seguimiento por nefrología, y en tratamiento con ACO desde hace 2 años. Intervenida en 2013 mediante laparoscopia por mioma uterino con diagnóstico anatomopatológico de leiomioma. Fue remitida a consultas ante el hallazgo causal en la TC de control por poliquistosis renal de 2 masas de 6,3 y de 3,1cm localizadas en espacio de Morrison y en mesosigma, sin poder descartar implantes peritoneales. La paciente se encontraba asintomática, y la exploración abdominal era normal. Se realizó PAAF de ambas lesiones, informando de neoplasia mesenquimal de bajo índice de proliferación, que expresaban receptores estrogénicos, Bcl-2 y vimentina, compatible con leiomioma entre otros diagnósticos, siendo imprescindible su exéresis para una correcta filiación. Se completó estudio con PET/TC (fig. 1A) que mostraba una intensa captación de ambas lesiones, con SUV máximo de 26,4 y 24,8, además de un útero aumentado de tamaño de aspecto miomatoso.
 PET/TC mostrando 2 focos de captación a nivel de las lesiones en espacio de Morrison y mesosigma con SUV máximo de 26,4 y 24,8, respectivamente. B) Pieza quirúrgica de la lesión localizada en espacio de Morrison.")
La paciente fue intervenida quirúrgicamente mediante una laparotomía media, identificando las lesiones y realizando resección completa de ambas (fig. 1B). La anatomía patológica informaba de neoplasias mesenquimales de bajo potencial maligno (Ki-67 al 2%, <1 mitosis/10CGA) con inmunofenotipo de músculo liso y receptores estrogénicos positivos, compatibles con leiomiomas. Se compararon los resultados, tanto de hematoxilina-eosina como de inmunohistoquímica, con la miomectomía previa realizada en 2013, siendo muy similares ambas lesiones, con la diferencia que las actuales presentaban una mayor vascularización y una menor positividad para desmina. Ante estos resultados, llamaba la atención la discrepancia entre los altos valores del SUV del PET/TC y el aspecto de bajo grado histológico (escasez de mitosis y ausencia de signos de malignidad como pleomorfismo y necrosis), por lo que se remitió a ginecología para valoración de histerectomía y doble anexectomía.
A los 3 meses la paciente fue intervenida por ginecología realizando histerectomía abdominal con doble anexectomía, hallando durante la intervención una nueva lesión de 1cm adherida a peritoneo parietal, con los mismos resultados anatomopatológicos que las lesiones resecadas previamente, aunque con un Ki-67 del 35%, sin evidenciar hallazgos patológicos en la pieza de histerectomía con doble anexectomía. Fue remitida a oncología, iniciando tratamiento hormonal adyuvante con tamoxifeno, sin signos de recidiva en la TC de control a los 6meses.
Aunque normalmente se trata de una enfermedad benigna, se ha descrito progresión hacia malignidad en un 3-5% de los casos3. El diagnóstico diferencial se establece principalmente con la leiomiosarcomatosis, la carcinomatosis peritoneal y los linfomas.
Microscópicamente son neoplasias mesenquimales constituidas por fibras musculares lisas, con presencia de receptores hormonales estrogénicos y de progesterona1,2, aunque los niveles hormonales del paciente son normales en la mayoría de casos. Por todo ello, se considera que la predisposición individual constituye un factor muy importante en el desarrollo de la enfermedad3.
Debido al escaso número de casos descritos, el tratamiento revisado en la bibliografía es variable1,3, en función de las características del paciente, desde actitud conservadora con seguimiento clínico-radiológico, hasta la cirugía radical (histerectomía con doble anexectomía y extirpación de todas las lesiones) con el fin de disminuir el influjo hormonal, evitando así la posibilidad de degeneración maligna. En caso de tumores residuales, puede ser útil un tratamiento hormonal adyuvante. El tratamiento con quimioterapia se reserva para aquellos casos que presenten degeneración maligna.

