




Sr. Director: La enfermedad de Whipple es una enfermedad sistémi ca crónica, causada por un actinomiceto grampositivo denominado Tropheryma whippelii y que puede afectar a casi cualquier órgano, pero de forma predominante al intestino delgado, el sistema nervioso central (SNC) y las articulaciones. Las manifestaciones oculares aparecen aproximadamente en un 5% de los pacientes con esta enfermedad, en forma de uveítis, vitritis, retinitis, neuritis retrobulbar, afectación directa del cristalino y glaucoma, entre otras1. Presentamos el caso de un paciente con enfermedad de Whipple que durante su evolución presentó una panuveítis.
Varón de 65 años de edad, minero de profesión, fumador de 2 paquetes al día, bebedor excesivo habitual (> 80 g/día), con diarrea (10-12 deposiciones en el día) de 2 meses de evolución, de características pastosas, sin sangre ni productos patológicos, que no respeta el sueño, con una pérdida de 6 kg de peso en este tiempo y artralgias en ambos hombros. En la exploración física destaca su delgadez (46 kg, 170 cm de altura); no hay adenopatías periféricas y sí un mínimo empastamiento maleolar bilateral; el resto de la exploración fue irrelevante. Entre las pruebas complementarias realizadas, presenta una anemia microcítica (hemoglobina 9,1 g/dl, hematocrito 28%, VCM 79 fl); leucocitos y plaquetas normales; velocidad de sedimentación globular 50 mm/h (0-15), proteína C reactiva 3,8 mg/dl (0-0,5). Bioquímica: GOT 47 U/l, FA 133 U/l, PT 6 g/dl; iones, calcio, vitamina D y resto de vitaminas liposolubles normales. Estudio de coagulación normal. Estudio en fresco de heces y co-procultivo negativo. La serología del virus de la inmunodeficiencia humana, virus de las hepatitis A, B y C, virus de Epstein-Barr y citomegalovirus fue negativa. Autoinmunidad (anticuerpos antinucleares), anticuerpos antitransglutaminasa, hormonas tiroideas y marcadores tumorales (CEA) negativos. Vitamina B12 y ácido fólico normales. El tránsito intestinal era compatible con un proceso malabsortivo (incremento difuso del calibre de las asas del intestino delgado, con signos de hipotonía y notorio enlentecimiento del tránsito, floculación y fragmentación de la columna de bario). En la endoscopia oral, el duodeno aparece con pliegues engrosados y un punteado blanquecino de tipo miliar. La biopsia duodenal muestra unas vellosidades atróficas y una lámina propia expandida y repleta de macrófagos con abundante material PAS+/amilasa resistente; la tinción de Ziehl no muestra micobacterias, compatible con enfermedad de Whipple (figs. 1 y 2).
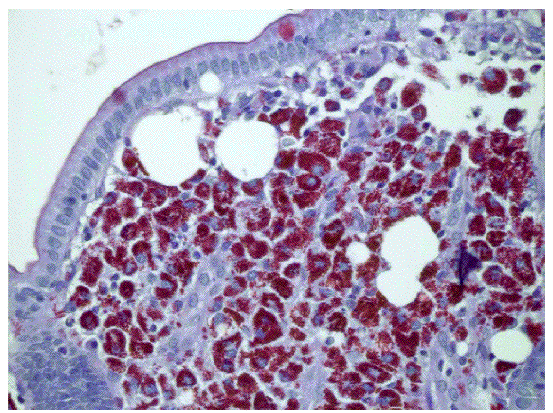
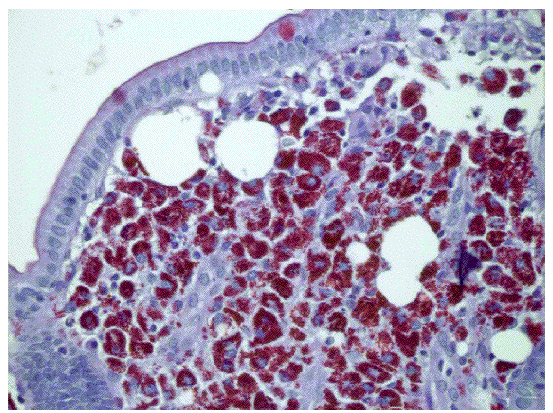
Fig. 1. Tinción con amilasa. Se aprecian acumulaciones de macrófagos espumosos en la mucosa intestinal.
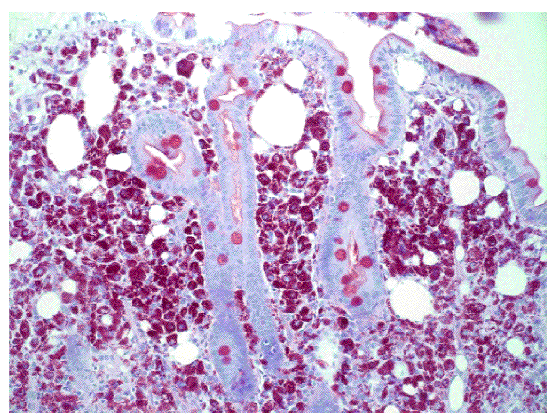
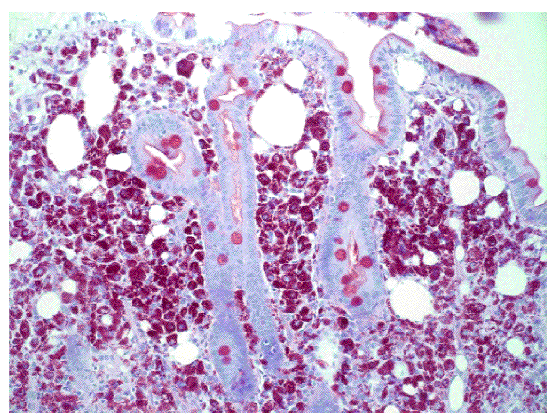
Fig. 2. Tinción con PAS. También se aprecian acumulaciones de macrófagos espumosos en la mucosa intestinal.
Inicialmente, el paciente se mostró reticente al tratamiento, por larga duración, lo que, en el tiempo de espera, le llevó a sufrir una panuveítis izquierda, estudiada y tratada por los oftalmólogos mediante corticoides tópicos, con buena resolución. Finalmente, tras informarle acerca del tratamiento, se inició la administración por vía oral de trimetoprim-sulfametoxazol en las dosis recomendadas (160/800 mg/12 h) durante un año, tras un ciclo de ceftriaxona previo (2 g i.v.) durante 2 semanas. El paciente ha sido seguido de forma ambulatoria posteriormente, y evolucionó de forma favorable, quedando asintomático, sin diarrea, sin anemia y con una ganancia ponderal de 5 kg; no obstante, en la endoscopia de control realizada tras un año del inicio del tratamiento, aún persiste la biopsia intestinal positiva para T. whippelii.
La enfermedad de Whipple es una enfermedad infrecuente. Aunque su fisiopatología no es bien conocida, parece haber 2 factores interrelacionados entre sí que contribuyen a su desarrollo: el agente infeccioso (T. whippelii) y el sistema inmunitario del huésped. Se ha observado un aumento del antígeno HLA-B27 en los pacientes afectados, así como una disminución del cociente CD4/CD8 en la lámina propia por incremento de la subpoblación CD8, y una disminución de la producción de interleucina (IL) 12 y de las células plasmáticas que contienen inmunoglobulina (Ig) A2. Es más frecuente en varones (8:1) de raza blanca, pero puede aparecer a cualquier edad, aunque normalmente se inicia en torno a los 50 años. La forma de presentación puede ser muy variada, según los sistemas o aparatos afectados. Aunque la tríada clínica más característica la constituye la aparición de fiebre, diarrea y artralgias, los signos y síntomas de la enfermedad pueden ser poco específicos, y varía ampliamente su forma de presentación, lo que puede hacer difícil su reconocimiento. Las manifestaciones oculares (panuveítis en nuestro paciente) aparecen aproximadamente entre un 5-15% de los pacientes. En general, la visión es completa o se produce una leve-moderada disminución de la agudeza visual. La mayoría de los pacientes tienen afectado el SNC o gastrointestinal y, de forma concomitante, aparecen síntomas sistémicos. Las manifestaciones oculares pueden derivarse de la enfermedad neurológica o de la afectación directa ocular (como en el caso presentado), y esta última es la situación más infrecuente3.
Clínicamente, el diagnóstico está basado en la demostración de macrófagos PAS+ en el tejido afectado, hallazgo indicativo pero no patognomónico, pues pueden visualizarse en otras enfermedades, como la histoplasmosis diseminada o la macroglobulinemia, y en los pacientes con sida e infección por Mycobacterium avium-intracellulare4. En sentido estricto, sería necesaria para el diagnóstico la demostración de los bacilos mediante microscopia electrónica o material genético de estos mediante ciertas técnicas, como la reacción en cadena de la polimerasa, que efectiva además en la monitorización de la respuesta al tratamiento. Actualmente, el tratamiento de elección es el trimetoprim-sulfametoxazol en dosis de 160/800 mg/12 h por vía oral durante al menos un año, comenzando con una fase de inducción las primeras 2 semanas con 1,2 millones de unidades al día de bencilpenicilina, asociada a 1 g de estreptomicina al día por vía parenteral, o una cefalosporina de tercera generación, como ceftriaxona5. Se aconseja realizar un seguimiento clínico del paciente de al menos 10 años para evitar las posibles recaídas. No hay un acuerdo claro sobre si es preciso realizar una biopsia intestinal de control tras finalizar el tratamiento. Algunos autores aconsejan rebiopsiar a los 3 meses de iniciado el tratamiento y, posteriormente, una vez al año durante 5 años. No obstante, para otros, la actitud más adecuada es repetir la biopsia únicamente en el caso de que el paciente no responda al tratamiento o si hay una recaída clínica6.

