




Sr. Director:
El síndrome de Lynch constituye la forma más frecuente de cáncer colorrectal (CCR) hereditario (0,9-2%). Tiene un patrón de herencia autosómico-dominante con alta penetrancia (80%), y se caracteriza por el desarrollo temprano de CCR, con características distintivas con respecto al CCR esporádico (más frecuente en colon derecho, neoplasias sincrónicas o metacrónicas e infiltración linfocitaria). El síndrome de Lynch se asocia a tumores en otras localizaciones (endometrio, ovario, estómago, intestino delgado, vías biliares, páncreas, vías urinarias, piel y cerebro). Desde el punto de vista patogénico existe una alteración del sistema de reparación del ADN (compuesto por los genes MLH1, MSH2, MSH6 y PMS2), acumulándose mutaciones en fragmentos de ADN altamente repetitivos, denominados microsatélites. Por otro lado, los tumores de estos pacientes pierden la expresión de la proteína correspondiente al gen mutado a nivel germinal (detectable mediante inmunohistoquímica). La estrategia para identificar los pacientes con síndrome de Lynch se basa en el cumplimiento de criterios clínicos (criterios revisados de Bethesda) y/o la realización de estudio del sistema de reparación del ADN en el tumor (mediante inmunohistoquímica para las proteínas reparadoras del ADN o estudio de inestabilidad de microsatélites). Cuando estas alteraciones están presentes se debe confirmar la sospecha mediante el estudio de la mutación germinal en los genes reparadores del ADN en sangre periférica. La identificación de la mutación causante permite ofrecer el estudio genético a los familiares de primer grado de los portadores de la mutación. La mayor parte de los casos de deben a mutaciones en los genes MSH2 (38%) y MLH1 (59%), y una pequeña proporción en MSH6 y PMS21,2.
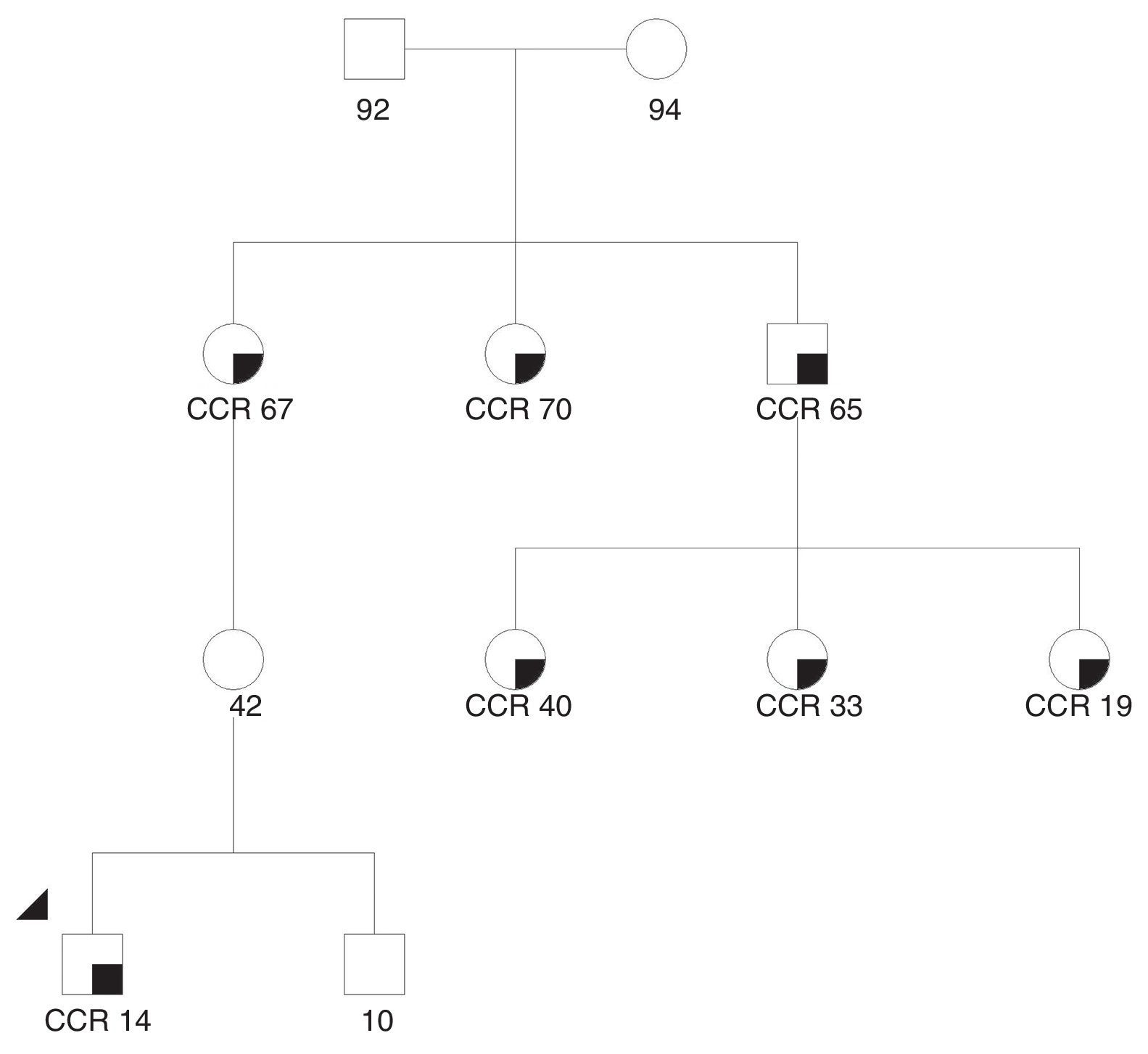
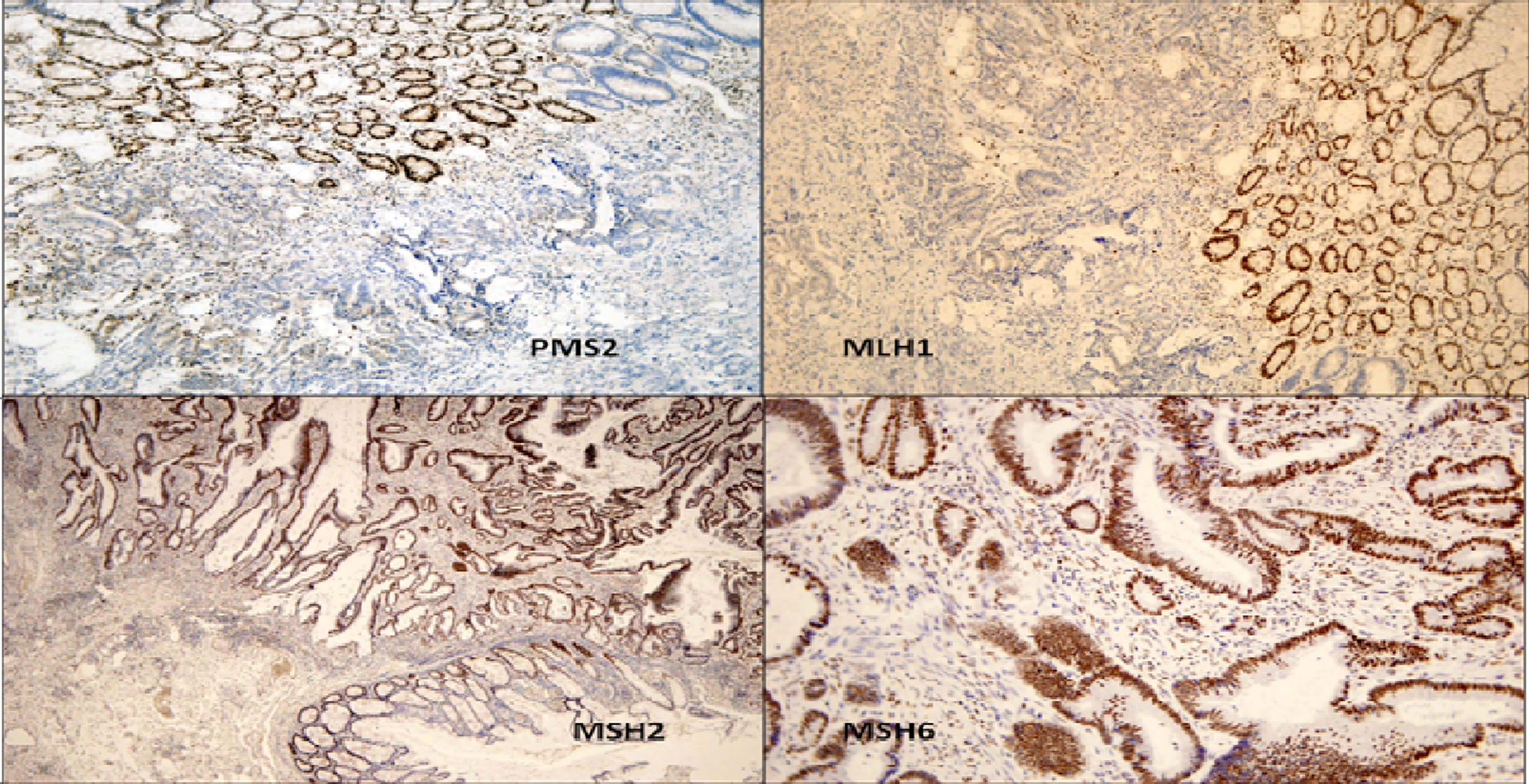
Presentamos el caso de un varón de 14 años de edad, de nacionalidad paraguaya, con antecedentes familiares de neoplasia colónica (fig. 1): abuela y 2 hermanos de esta lo presentaron. Ingresa por diarrea crónica con restos hemáticos. En la colonoscopia se visualiza un tumor en recto proximal, que se biopsia, resultando un adenocarcinoma. El estudio de extensión realizado con RMN y TAC, delimitó una masa en 6×6cm, infiltrando la pared posterior de la vejiga, sin evidencia de metástasis. El paciente fue sometido a radio-quimioterapia neoadyuvante, y posteriormente intervenido quirúrgicamente, practicándose escisión total del mesorrecto y cara posterior de vejiga. La pieza macroscópica mostró adenocarcinoma ulcerado e infiltrante que invadía la muscular externa vesical, con diferenciación mucosecretora, con fascia mesorrectal íntegra, 12 ganglios linfáticos negativos, y ausencia de invasión vascular y perineural. El paciente realizó tratamiento adyuvante con oxaliplatino y capecitabina. Se solicitó estudio inmunohistoquímico para MLH1/MSH2/MSH6/PMS2 en el tumor del paciente, detectándose pérdida de expresión de las proteínas MLH1 y PMS2 en las células tumorales (fig. 2). No se realizó estudio genético confirmatorio de síndrome de Lynch al trasladarse a su país de origen. Tras 58 meses de seguimiento se halla libre de enfermedad. Hasta el momento actual, 3 hijas de un tío-abuelo han sido diagnosticadas de CCR, todas con edades inferiores a 40 años.


Estudio inmunohistoquímico para detectar la expresión de las proteínas asociadas a los genes reparadores del DNA: se observa pérdida de expresión de PMS2 y MLH1 en las células tumorales, mientras que la mucosa normal conserva la expresión nuclear de las 2 proteínas. MSH2 y MSH6 son positivos en el tumor y en la mucosa normal.
El diagnóstico del síndrome de Lynch supone un reto en la práctica clínica. Tras el diagnóstico de sospecha basado en la clínica y el estudio del sistema de reparación del ADN en el tumor, la presencia de mutaciones germinales en los genes reparadores del ADN, nos permiten confirmar el diagnóstico, y realizar el cribado del mismo en los familiares en riesgo3. Sin embargo, el análisis genético está poco introducido en la práctica clínica habitual, y su interpretación puede resultar compleja. Por ello se aconseja el estudio y seguimiento de estos pacientes en unidades de consejo genético especializadas. El cribado endoscópico (colonoscopias cada 1-2 años) puede centrarse únicamente en los miembros portadores de la mutación causal, dirigido a la identificación de lesiones precursoras o carcinomas en fases iniciales. La cromoendoscopia ha demostrado aumentar la detección de lesiones en pacientes con síndrome de Lynch4.
No hemos hallado descrito, en la literatura, ningún caso de CCR hereditario con 2 pacientes tan jóvenes padeciendo la enfermedad. En el caso expuesto, podría plantearse el diagnóstico diferencial con el síndrome de deficiencia del sistema reparador constitucional, definido por la presencia de mutaciones bialélicas en los genes reparadores del ADN. Sin embargo, la ausencia de casos en la rama paterna, la presencia de expresión normal de MLH1/PMS2 en la mucosa normal, y la ausencia de otras manifestaciones (manchas café con leche, tumores hematológicos) hacen improbable su presencia5. Por ello, en esta familia, pensamos que el diagnóstico más probable es el síndrome de Lynch por mutación en MLH1. De confirmarse el diagnóstico de sospecha, sería recomendable iniciar el cribado endoscópico a los 10 años de edad en los familiares en riesgo. Asimismo, se recomienda realizar cribado de neoplasia de endometrio y de neoplasias extra-colónicas en función de la predisposición familiar a un determinado tumor. El tratamiento de los tumores colorrectales en estos pacientes es la resección quirúrgica extensa para prevenir neoplasias metacrónicas, con seguimiento endoscópico posterior cada 1-2 años. Los tumores de los pacientes con síndrome de Lynch se asocian a un mejor pronóstico, hecho que podría explicar la supervivencia libre de enfermedad tan prolongada, pese a tratarse de un tumor localmente avanzado.

