




La obesidad se asocia a un mayor riesgo de tener una enfermedad hepática por depósito de grasa no relacionada con el abuso de alcohol (EHGNA) y contribuye a la progresión de hepatopatías de diferentes etiologías, como la hepatitis crónica por el virus de hepatitis C (VHC). El descubrimiento de que el tejido adiposo es un tejido sometido a un estado de inflamación crónica capaz de secretar adipoquinas ha permitido establecer un nexo de unión entre las alteraciones metabólicas que conducen al acúmulo de triglicéridos y a la inflamación hepática, y ha reforzado el papel de la lipotoxicidad hepatocelular en la patogenia de la enfermedad hepática por depósito de grasa no relacionada con el abuso de alcohol. Por otro lado, aunque el genotipo 3 del VHC induce esteatosis, actualmente se considera que la obesidad y sus alteraciones metabólicas asociadas, como la resistencia a la insulina, están implicadas en la progresión de la enfermedad hepática mediada por el VHC así como de otras hepatopatías crónicas de diversas etiologías.
Obesity is associated with a higher risk of developing non-alcoholic fatty liver disease (NAFLD) and contributes to the progression of liver diseases of distinct etiologies such as chronic hepatitis C virus (HCV) infection. The discovery that adipose tissue is submitted to a state of chronic inflammation able to secrete adipokines has allowed a connection to be established between the metabolic alterations that lead to triglyceride accumulation and liver inflammation, reinforcing the role of hepatocellular lipotoxicity in the pathogenesis of NAFLD. In addition, although HCV genotype 3 induces steatosis, it is currently believed that obesity and its associated alterations, such as insulin resistance, are involved in progression of HCV-mediated liver disease, as well as that of other chronic liver diseases of diverse etiologies.
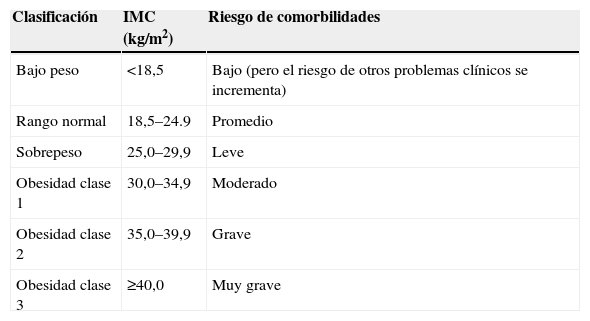
La obesidad es una enfermedad crónica que suele iniciarse en la infancia o en la adolescencia y que se produce como consecuencia de un desequilibrio entre la ingesta y el gasto energético. En su origen se involucran factores genéticos y ambientales, que determinan un trastorno metabólico sistémico que conduce a una excesiva acumulación de grasa corporal. En la práctica clínica actual, los criterios más utilizados para el diagnóstico de sobrepeso y obesidad, ya sea en niños o en adultos, están basados en el índice de masa corporal (IMC). En la tabla 1 se exponen los límites de IMC acordados y establecidos por la Organización Mundial de la Salud, que constituyen la clasificación diagnóstica de obesidad más ampliamente aceptada.
Clasificación de la obesidad según la Organización Mundial de la Salud
| Clasificación | IMC (kg/m2) | Riesgo de comorbilidades |
| Bajo peso | <18,5 | Bajo (pero el riesgo de otros problemas clínicos se incrementa) |
| Rango normal | 18,5–24.9 | Promedio |
| Sobrepeso | 25,0–29,9 | Leve |
| Obesidad clase 1 | 30,0–34,9 | Moderado |
| Obesidad clase 2 | 35,0–39,9 | Grave |
| Obesidad clase 3 | ≥40,0 | Muy grave |
IMC: índice de masa corporal.
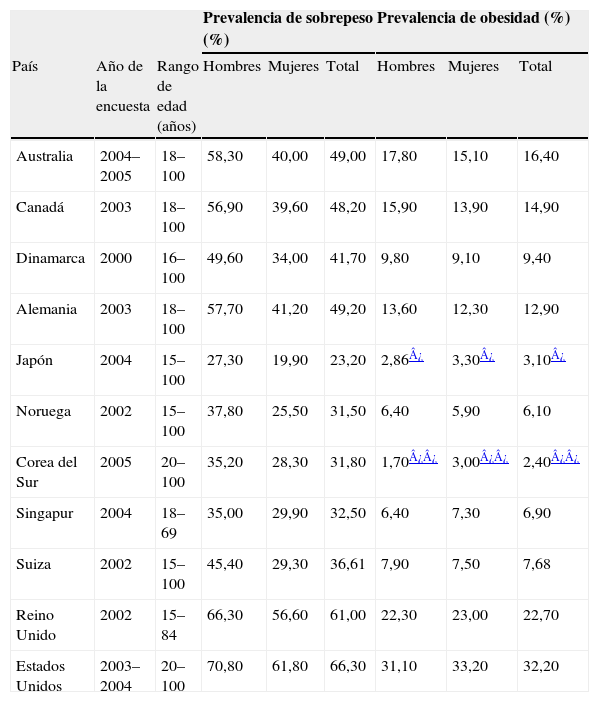
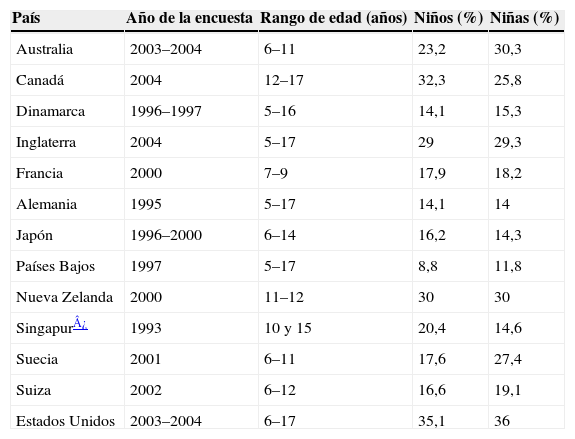
Estudios epidemiológicos realizados en distintos países muestran que el 5–10% de los niños en edad escolar son obesos, en la población adolescente la proporción aumenta hasta el 10–20% y en los adultos las cifras de prevalencia de obesidad oscilan entre el 7,7% en Suiza y el 32,2% en Estados Unidos1,2 (tablas 2 y 3). Más elocuentes aún son las cifras aportadas por la Organización Mundial de la Salud, que estima en 400 millones el número de personas adultas obesas en el año 2005, al tiempo que pronostica que en el año 2015 habrá alrededor de 700 millones de adultos obesos en el mundo3, lo que indica que la obesidad representa en la actualidad un problema sanitario mundial de primer orden.
Prevalencia en países desarrollados de sobrepeso y obesidad en adultos
| Prevalencia de sobrepeso (%) | Prevalencia de obesidad (%) | |||||||
| País | Año de la encuesta | Rango de edad (años) | Hombres | Mujeres | Total | Hombres | Mujeres | Total |
| Australia | 2004–2005 | 18–100 | 58,30 | 40,00 | 49,00 | 17,80 | 15,10 | 16,40 |
| Canadá | 2003 | 18–100 | 56,90 | 39,60 | 48,20 | 15,90 | 13,90 | 14,90 |
| Dinamarca | 2000 | 16–100 | 49,60 | 34,00 | 41,70 | 9,80 | 9,10 | 9,40 |
| Alemania | 2003 | 18–100 | 57,70 | 41,20 | 49,20 | 13,60 | 12,30 | 12,90 |
| Japón | 2004 | 15–100 | 27,30 | 19,90 | 23,20 | 2,86¿ | 3,30¿ | 3,10¿ |
| Noruega | 2002 | 15–100 | 37,80 | 25,50 | 31,50 | 6,40 | 5,90 | 6,10 |
| Corea del Sur | 2005 | 20–100 | 35,20 | 28,30 | 31,80 | 1,70¿¿ | 3,00¿¿ | 2,40¿¿ |
| Singapur | 2004 | 18–69 | 35,00 | 29,90 | 32,50 | 6,40 | 7,30 | 6,90 |
| Suiza | 2002 | 15–100 | 45,40 | 29,30 | 36,61 | 7,90 | 7,50 | 7,68 |
| Reino Unido | 2002 | 15–84 | 66,30 | 56,60 | 61,00 | 22,30 | 23,00 | 22,70 |
| Estados Unidos | 2003–2004 | 20–100 | 70,80 | 61,80 | 66,30 | 31,10 | 33,20 | 32,20 |
Prevalencia en países desarrollados de sobrepeso en la infancia (incluyendo la obesidad)
| País | Año de la encuesta | Rango de edad (años) | Niños (%) | Niñas (%) |
| Australia | 2003–2004 | 6–11 | 23,2 | 30,3 |
| Canadá | 2004 | 12–17 | 32,3 | 25,8 |
| Dinamarca | 1996–1997 | 5–16 | 14,1 | 15,3 |
| Inglaterra | 2004 | 5–17 | 29 | 29,3 |
| Francia | 2000 | 7–9 | 17,9 | 18,2 |
| Alemania | 1995 | 5–17 | 14,1 | 14 |
| Japón | 1996–2000 | 6–14 | 16,2 | 14,3 |
| Países Bajos | 1997 | 5–17 | 8,8 | 11,8 |
| Nueva Zelanda | 2000 | 11–12 | 30 | 30 |
| Singapur¿ | 1993 | 10 y 15 | 20,4 | 14,6 |
| Suecia | 2001 | 6–11 | 17,6 | 27,4 |
| Suiza | 2002 | 6–12 | 16,6 | 19,1 |
| Estados Unidos | 2003–2004 | 6–17 | 35,1 | 36 |
En los últimos años, la evidencia clínica y epidemiológica ha puesto de manifiesto que la obesidad, además de ser un factor común de riesgo para diversas enfermedades, como la diabetes, la enfermedad cardiovascular y determinados tipos de cáncer, se asocia a un mayor riesgo de presentar una enfermedad hepática por depósito de grasa no relacionada con el abuso de alcohol (EHGNA) y contribuye a la progresión de hepatopatías de diferentes etiologías, como la hepatitis crónica por el virus de la hepatitis C (VHC). En la presente revisión se hará especial énfasis en la evidencia clínica y epidemiológica existente que relaciona la obesidad con los trastornos del hígado y en los mecanismos patogénicos de la enfermedad hepática asociada a la obesidad.
La obesidad se asocia a trastornos del hígadoEn el clásico estudio Dyonisos4, que se llevó a cabo en una población del norte de Italia, se observó que el 76% de las personas obesas no alcohólicas y alrededor del 15% de las personas no obesas tenían evidencia ecográfica de hígado graso. En estudios posteriores, no obstante, se comprobó que la prevalencia de hígado graso varía considerablemente en función del método diagnóstico utilizado, de la raza y del sexo. Así, en un estudio realizado en 2.287 habitantes de diferentes ciudades estadounidenses, a los que se les determinó el contenido hepático de triglicéridos mediante un sofisticado método de espectroscopia protónica por resonancia magnética, se observó que aproximadamente un tercio de la población tenía esteatosis hepática y que ésta era más frecuente en las personas de origen latinoamericano (45%) que en las de raza blanca (33%) o negra (24%), así como en los hombres (42%) con respecto a las mujeres (24%). Un hallazgo interesante de este estudio fue que la mayor frecuencia de esteatosis en la población hispana se debía a una mayor prevalencia de obesidad5. Una tendencia similar en relación con la mayor prevalencia de esteatosis en hombres que en mujeres se ha observado en poblaciones de origen asiático6, así como que pequeñas variaciones en el peso corporal (entre 1,3–2,5kg de promedio) se asocian a variaciones significativas en el patrón ecográfico de esteatosis hepática7, lo que indica que el grado de obesidad influye en la acumulación de grasa en el hígado.
Algunos estudios epidemiológicos han indicado que el aumento de las concentraciones séricas de las enzimas hepáticas es un indicador sensible de esteatosis hepática4,8, y es por esto que se han utilizado en estudios poblacionales como un marcador de daño hepático. Con esta estrategia, diferentes estudios poblaciones realizados en distintas áreas geográficas han observado que cuanto mayor es el IMC más alta es la prevalencia de enzimas hepáticas elevadas y que la presencia de obesidad visceral, medida por el cociente entre el perímetro de la cintura y el de la cadera, es un factor determinante de la significativa asociación entre la obesidad y el aumento de las concentraciones séricas de aminotransferasas9,10.
La obesidad no sólo se ha relacionado con las fases iniciales de la EHGNA, sino además con el riesgo de progresar a esteatohepatitis y también a cirrosis y carcinoma hepatocelular (CHC). En este sentido, un estudio realizado en Estados Unidos y basado en la First National Health and Nutrition Examination Survey comprobó que las hospitalizaciones o las muertes relacionadas con cirrosis hepática eran más frecuentes en las personas obesas (0,81/1000 personas por año) y en las que tenían sobrepeso (0,71/1000 personas por año) que en aquéllas con peso normal (0,45/1000 personas por año), independientemente de la ingesta de alcohol11. Por otro lado, en un estudio poblacional prospectivo en el que se evaluaron 900.000 adultos estadounidenses que no tenían cáncer al inicio del estudio en 1982 y a los que se siguió durante 16 años de media, se demostró, tanto en hombres como en mujeres, una correlación positiva entre el IMC y la muerte por distintos tipos de cáncer. En concreto, un IMC superior a 35kg/m2 se asociaba a un riesgo relativo de muerte por CHC de 4,52 con respecto a las personas con IMC normal12. En la misma línea, en un estudio realizado en pacientes trasplantados por CHC también se observó que la obesidad era un factor predictivo independiente de CHC en pacientes con cirrosis alcohólica y en pacientes con cirrosis criptogénica. No está clara la asociación patogénica entre la obesidad y el CHC, pero la evidencia experimental procedente de modelos animales de obesidad indica que las alteraciones metabólicas relacionadas con la obesidad, más que la cirrosis per se, podrían estar implicadas en la inducción de hepatocarcinogénesis durante la obesidad13.
La obesidad es un factor etiológico del hígado graso no alcohólicoLa revisión detallada de los grandes estudios epidemiológicos poblacionales nos indica que la obesidad aumenta en 2 o 3 veces el riesgo de tener concentraciones séricas elevadas de enzimas hepáticas, mientras que el riesgo de esteatosis ecográfica aumenta 3 veces en la personas con sobrepeso y hasta 15 veces en presencia de obesidad14. Todos estos datos se han visto confirmados en diferentes estudios realizados en cohortes de pacientes con obesidad mórbida15–18, en los que se ha observado que la mayoría de estos pacientes tiene esteatosis hepática (91%, rango: 85–98%), alrededor de un tercio tiene signos histológicos de esteatohepatitis (37%, rango: 24–98%), de los cuales el 20–40% presenta un estadio avanzado de fibrosis e incluso cirrosis en alrededor del 2%. El concepto de que la obesidad es un factor etiológico de la EHGNA se ha visto reforzado recientemente a la vista de los resultados obtenidos en estudios longitudinales de cohortes de pacientes con obesidad mórbida, a los que se ha estudiado metabólica e histológicamente tras la realización de cirugía bariátrica. La reducción de peso en estos pacientes se correlacionaba con la mejoría de las alteraciones metabólicas, como la resistencia a la insulina (RI) y las concentraciones séricas de adipoquinas, así como de las lesiones histológicas hepáticas características de la EHGNA, como la esteatosis, la degeneración balonizante y la fibrosis19–21.
Aunque se considera que la EHGNA es una enfermedad hepática de evolución lentamente progresiva con respecto a otras hepatopatías crónicas, la probabilidad de evolucionar a cirrosis es un hecho cada vez más reconocido22. En este sentido, diferentes estudios han evaluado la presencia de factores de riesgo metabólico en cohortes de pacientes cirróticos y han encontrado que la prevalencia de obesidad o diabetes, o ambas, en los pacientes con cirrosis criptogénica era similar a la de pacientes con esteatohepatitis no alcohólica (EHNA), pero significativamente más alta que en los pacientes con cirrosis de etiología vírica o alcohólica, lo que indica que la cirrosis criptogénica puede representar el estadio evolutivo final de la EHNA23–25. El hecho de que los pacientes con cirrosis criptogénica desarrollen con frecuencia obesidad y EGHNA postrasplante apoya el concepto de que la obesidad es un factor etiológico de la EGHNA y que ésta puede evolucionar potencialmente hacia formas avanzadas de enfermedad hepática como la cirrosis26. Existe además evidencia clínica que indica que el riesgo de CHC en pacientes con cirrosis asociada a EHGNA es similar al de pacientes con cirrosis alcohólica o por infección crónica por el VHC27. Más concretamente, en otro estudio restrospectivo se observó que el 27% de los pacientes obesos con cirrosis criptogénica tenía CHC en comparación con el 21% de los cirróticos por VHC, lo que indica que el potencial carcinogénico de la obesidad es similar al del VHC en presencia de cirrosis28.
En la última década se han llevado a cabo numerosos estudios longitudinales, con biopsias hepáticas seriadas, cuyo principal objetivo ha sido evaluar la progresión de la fibrosis y los factores de riesgo asociados en cohortes de pacientes con EHGNA. Así, Teli et al29 observaron, tras un seguimiento medio de 11 años, que la progresión de la fibrosis era mucho más frecuente en los pacientes con esteatohepatitis (8%) que en aquéllos con esteatosis simple (0%). Uno de los factores que parece estar implicado en la progresión de la fibrosis es la obesidad, ya que en otro estudio longitudinal más reciente se puso de manifiesto que la obesidad era significativamente más prevalerte en los pacientes con fibrosis progresiva (86%) que en aquéllos en los que la fibrosis permanecía estable (27%)30. Además de la obesidad, la presencia de diabetes se ha relacionado con un mayor riesgo de progresión de la fibrosis en pacientes con EHGNA31.
Finalmente, existen pocos datos acerca del pronóstico a largo plazo de la EHGNA, pero en un estudio realizado en 129 pacientes, con un seguimiento medio de 13,7 años, se comprobó que los pacientes con esteatohepatitis tenían una tasa de mortalidad significativamente más alta que los pacientes con esteatosis simple, y que las causas de muerte más frecuentes son la enfermedad cardiovascular y la enfermedad hepática avanzada32.
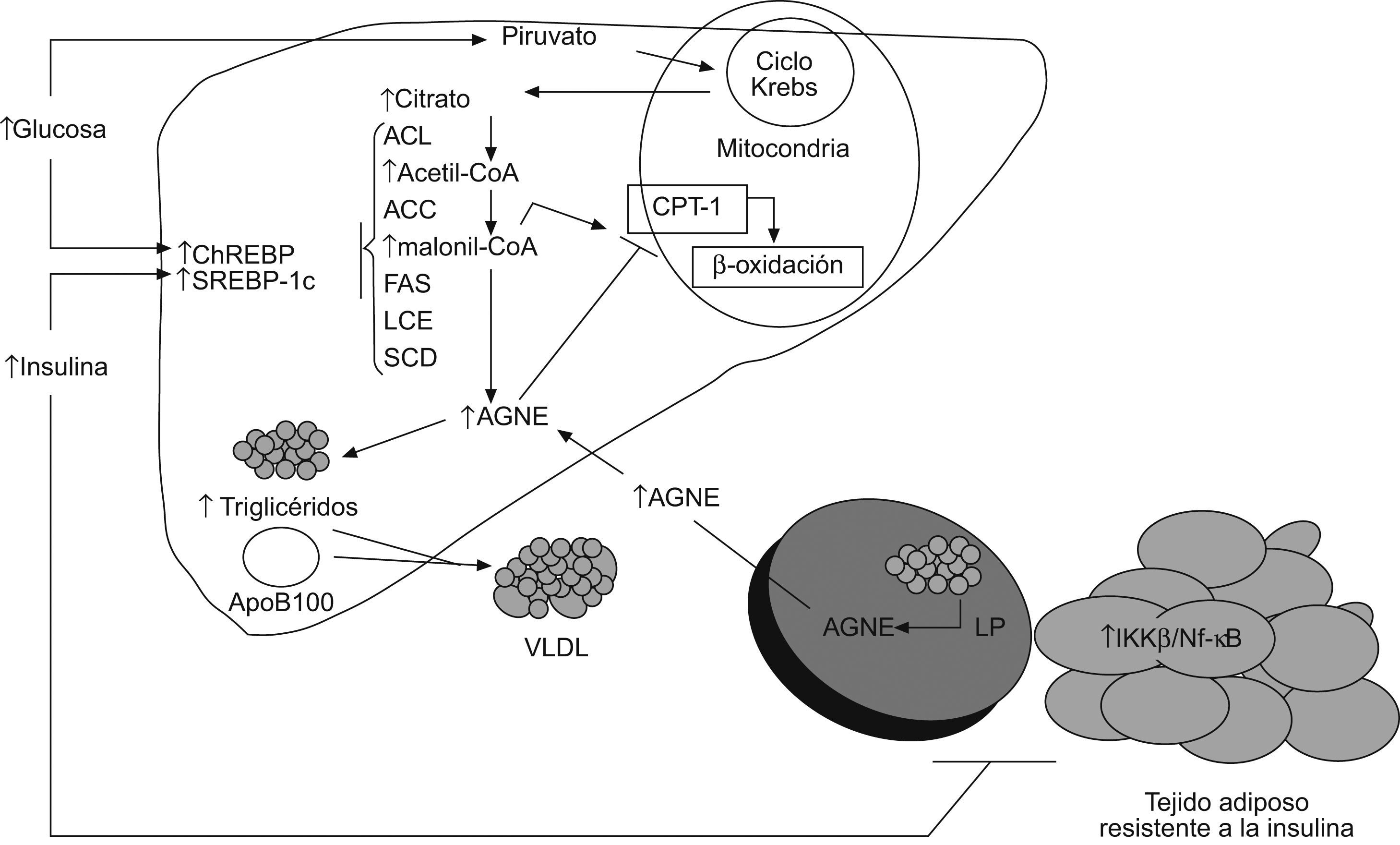
Patogenia del hígado graso no alcohólicoEn condiciones fisiológicas, la homeostasis lipídica requiere de la existencia de interacciones metabólicas coordinadas entre el hígado, el músculo y el tejido adiposo, ejercidas en gran medida por la acción reguladora de la insulina. El hígado tiene un papel central en el metabolismo de los lípidos: capta los ácidos grasos (AG) circulantes que proceden fundamentalmente del tejido adiposo y en menor medida de la absorción intestinal de la grasa de la dieta. Además, los hepatocitos pueden sintetizar los AG de novo. Una vez en el hígado, los AG tienen 2 destinos fundamentales: incorporarse a las vías de oxidación intracelular para generar ATP o esterificarse para convertirse en triglicéridos y así secretarse a la sangre unidos a la apoliproteína B100 y formar lipoproteínas de muy baja densidad. Por tanto, aquellos procesos que aumenten la captación hepática de AG o alteren su metabolismo (síntesis, oxidación o esterificación) y su posterior secreción pueden producir un acúmulo de grasa en el hígado, el primer «impacto» en el modelo patogénico de la EHGNA propuesto por Cristopher P. Day y Oliver W. James en 199833, conocido como la teoría del doble «impacto».
Actualmente se considera que la RI constituye el pilar patogénico básico de la esteatosis hepática, ya que puede interferir el metabolismo hepático de los AG a diferentes niveles (fig. 1). Se ha descrito que la RI produce un aumento del flujo de AG no esterificados (AGNE) al hígado debido al incremento de la hidrólisis de los triglicéridos por una activación mantenida de la lipasa adipocitaria34. Además, la RI se acompaña de una desregulación de la tasa de captación hepática de AGNE, y en pacientes diabéticos se demuestra una relación directamente proporcional con las concentraciones séricas de AGNE35. La hiperinsulinemia y el aumento de la producción hepática de glucosa, que se producen como consecuencia de la RI, inducen la expresión de la proteína de unión al elemento regulador de esteroles (SREBP-1c) y de la proteína de unión al elemento de respuesta a hidratos de carbono (ChREBP), respectivamente, que a su vez activan la transcripción de la mayoría de los genes que participan en la maquinaria enzimática necesaria para la síntesis hepática (de novo) de AG a partir del exceso de hidratos de carbono36. Por otro lado, la SREBP-1c inhibe la transcripción del sustrato del receptor de la insulina-2, lo que induce o exacerba la RI a nivel hepático37.

Alteraciones metabólicas secundarias a la resistencia a la insulina que conducen al acúmulo de triglicéridos en el hígado. La inducción de la lipogénesis de novo, mediada por la insulina y la glucosa, la inhibición de la betaoxidación mitocondrial por los ácidos grasos no esterificados y por la malonil-CoA así como el aumento de la captación de ácidos grasos no esterificados circulantes contribuyen a la esteatosis hepática. ACC: acetil-CoA carboxilasa; ACL: ATP citrato liasa; AGNE: ácidos grasos no esterificados; ChREBP: proteína de unión al elemento de respuesta a hidratos de carbono; CPT-1: carnitina palmitoil transferasa-1; FAS: sintasa de ácidos grasos; IKK-β: cinasa β del inhibidor κB; LCE: elongasa de ácidos grasos de cadena larga; LP: lipasa insulinsensible; NF-κβ: factor nuclear κβ; SCD: estearoil-CoA desaturasa; SOCS: proteínas supresoras de la señalización de citoquinas; SREBP: proteína de unión al elemento regulador de esteroles; VLDL: lipoproteínas de muy baja densidad. →: vía estimuladora; ⊣: vía inhibitoria.
La teoría más clásica sobre la patogenia de la esteatosis hepática, denominada teoría portal, atribuye un papel clave al tejido adiposo visceral como fuente primordial de AGNE al hígado que circulan por la vena porta38. Esta teoría se ha visto reforzada con resultados provenientes de estudios clínicos y experimentales en pacientes con EHGNA, en los que se han observado concentraciones séricas elevadas de AGNE39 al tiempo que se ha demostrado que la mayoría (60%) del contenido intrahepático de triglicéridos proviene del pool circulante de los AGNE, mientras que un 25% procede de la síntesis de novo de AG y un 15% de los AG de la dieta40. A pesar de que faltan datos sobre la concentración de AGNE en la sangre portal de estos pacientes, hay suficiente evidencia para afirmar que son los AGNE circulantes que provienen de la hidrólisis del tejido adiposo los que contribuyen fundamentalmente a la esteatosis en los pacientes con EHGNA.
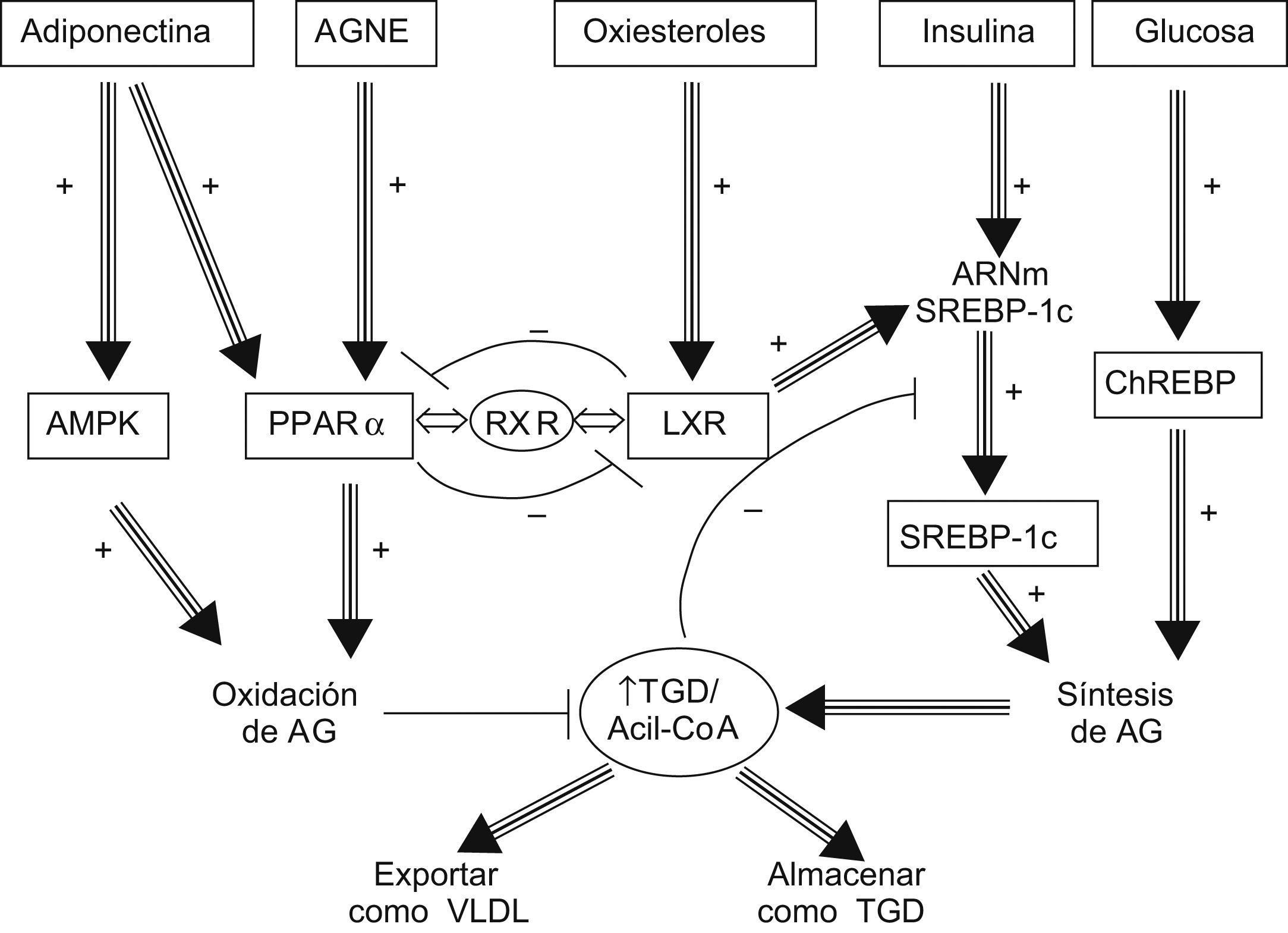
Independientemente de lo comentado hasta ahora, se han descrito otros factores que pueden contribuir al acúmulo de triglicéridos en el hígado, como la disminución de la betaoxidación mitocondrial41. En este sentido, se ha demostrado que la insulina inhibe esta vía fisiológica de oxidación hepática de AG al producir la activación, mediada por SREBP-1c, de la isoforma 2 de la acetil-CoA carboxilasa que produce malonil-CoA a nivel de la membrana mitocondrial42. El aumento de la síntesis de malonil-CoA disminuye la oxidación mitocondrial de los AG al inhibir la enzima carnitina palmitoiltransferasa que se encarga de transportar los AG de cadena larga desde el citoplasma al interior de la mitocondria43. El aumento de la lipogénesis hepática puede también contribuir, aunque de forma modesta como ya se ha mencionado, al acúmulo de triglicéridos. Se sabe que el metabolismo lipídico hepático está estrechamente regulado por una serie de moléculas mediadoras, como los receptores de los ligandos activadores de la proliferación peroxisomal (PPAR), el receptor X del hígado (LXR) y la proteína cinasa activada por AMP (AMPK)36.
Se conocen tres subtipos de PPAR: PPAR-α, PPAR-γ y PPAR-δ; este último se expresa casi exclusivamente en el músculo y tiene un papel menos relevante. El subtipo α se expresa fundamentalmente en los tejidos que utilizan los AG como fuente de energía, como el hígado, el músculo, el corazón y el riñón44. Cuando la concentración hepática de AG aumenta, el PPAR-α se activa y promueve la transcripción de genes implicados en la betaoxidación mitocondrial, peroxisomal y microsomal (acil-CoA oxidasa y citocromo P450 4A), así como en el transporte y la secreción de los AG del hígado (la proteína microsomal transportadora de triglicéridos y la apoliproteína B100)45,46. Por tanto, el resultado final de su activación es el incremento del catabolismo hepático de AG. El papel de PPAR-α en la patogenia de la EHGNA está mejor definido en ratones que en humanos. Hay evidencia de que la estimulación del PPAR-α tras la administración de agonistas a ratones alimentados con dieta deficiente en metionina y colina revierte la esteatohepatitis47. Por el contrario, el papel del PPAR-α en la patogenia de la EHGNA en humanos está menos claro. En este sentido, se ha comunicado que el polimorfismo L162V del gen del PPAR-α que se acompaña de una mayor actividad transcripcional, no se asocia a la presencia de EHGNA en humanos48. A diferencia del PPAR-α, el PPAR-γ se expresa fundamentalmente en los adipocitos, donde tiene un papel primordial en la lipogénesis y en la diferenciación normal de los adipocitos, y mejora así la sensibilidad a la insulina del tejido adiposo49,50. En condiciones fisiológicas, la expresión hepática del PPAR-γ es muy baja, mientras que en ratones con RI y esteatosis hepática la expresión hepática de este receptor nuclear está muy aumentada51,52. La importancia del PPAR-γ en la patogenia de la esteatosis hepática se puso de manifiesto cuando se demostró que la delección del gen del PPAR-γ en el hígado de estos animales se acompañaba de una franca mejoría de la esteatosis53. No obstante, los mecanismos moleculares por los que el PPAR-γ promueve el acúmulo hepático de triglicéridos no se conocen con exactitud.
Otro mediador relevante en la homeostasis hepática de las grasas es el LXR. Al igual que los PPAR, el LXR, tras su activación por parte de ciertos ligandos, forma complejos heterodiméricos con el receptor X retinoide que se comportan como transactivadores de la transcripción de genes implicados en la síntesis de AG, como el SREBP-1c y el ChREBP, lo que podría, por tanto, contribuir a la esteatosis54. El LXR interacciona con el PPAR-α de una manera recíprocamente inhibitoria, de modo que los 2 receptores ejercen funciones opuestas sobre el metabolismo lipídico. Tal como se representa en la figura 2, el LXR promueve la biosíntesis de los AG mientras que el PPAR-α induce la oxidación de éstos. La competición por el receptor X retinoide disponible en el citoplasma de la célula es el exclusivo mecanismo que regula la activación de uno u otro receptor, que polarizan a la célula hacia la síntesis o hacia el catabolismo lipídico55,56.

Homeostasis hepática de las grasas. El receptor X del hígado promueve la biosíntesis de los ácidos grasos mientras que el receptor α de los ligandos de la proliferación peroxisomal induce la oxidación de éstos. AGNE: ácidos grasos no esterificados; AMPK: quinasa activada por adenosin monofosfato; ChREBP: proteína de unión al elemento de respuesta a hidratos de carbono; LXR: receptor X del hígado; PPAR-α: receptor α de los ligandos de la proliferación peroxisomal; RXR: receptor X retinoide; SREBP: proteína de unión al elemento regulador de esteroles; TGD: triglicéridos.
La AMPK funciona como un sensor de los niveles energéticos de la célula57. Esta proteína cinasa se activa cuando aumentan los niveles intracelulares de AMP, lo que ocurre cuando disminuyen las reservas celulares de energía. La AMPK activada estimula las vías catabólicas de la célula que producen ATP, como la betaoxidación mitocondrial, e inhibe los procesos que consumen ATP, como la lipogénesis, directamente fosforilando proteínas reguladoras e indirectamente regulando la expresión de genes involucrados en estas vías metabólicas57. La composición de AG en el hepatocito puede modular la actividad de la AMPK. En este sentido, se ha demostrado que la delección genética de la enzima estearoil-CoA desaturasa, encargada de la síntesis de AG monoinsaturados, protege de la aparición de esteatosis hepática y de RI en ratones58,59. En ausencia de la enzima estearoil-CoA desaturasa, la AMPK se activa60 fosforilando e inhibiendo la acetil-CoA carboxilasa y la ChREBP61,62, así como disminuyendo los niveles de expresión de la SREBP-1c63. Las tiazolidindionas son fármacos antidiabéticos que se caracterizan por activar el PPAR-γ. Además, tanto estos fármacos como la metformina son capaces de activar la AMPK hepática63–65. Sus efectos beneficiosos en pacientes con EHGNA66–68 serían en parte la consecuencia de unos acontecimientos moleculares que básicamente tienen que ver con la activación de la vía de la AMPK.
En síntesis, el incremento de la lipogénesis hepática es una importante alteración metabólica que contribuye a la patogenia de la esteatosis hepática en pacientes con RI, aunque sólo el 25% de la grasa intrahepática en pacientes con EHGNA proviene de la síntesis de novo de AG. Hoy se considera que el incremento del flujo y de la captación hepática de AGNE circulantes procedentes de una lipólisis periférica excesiva, todo ello como consecuencia de la RI periférica, es el principal factor patogénico de la esteatosis hepática en humanos. Otros factores como la disminución de la betaoxidación mitocondrial así como de la secreción hepática de triglicéridos son menos importantes, pero pueden contribuir a la exacerbación del acúmulo de grasa en los hepatocitos.
Es un hecho conocido por estudios clínicos que sólo una proporción de pacientes con esteatosis hepática progresan a esteatohepatitis con o sin fibrosis69. El conocimiento de los mecanismos implicados en la progresión de una esteatosis simple, hasta ahora considerada como un trastorno hepático puramente metabólico, a una esteatohepatitis, el denominado «segundo impacto» patogénico, es un objetivo prioritario con el fin de diseñar estrategias más racionales para el tratamiento y la prevención de los pacientes que tienen o que están en riesgo de tener EHNA. Inicialmente se consideraron como los principales candidatos para este «segundo impacto» el estrés oxidativo, con la consiguiente peroxidación lipídica, y las citoquinas, fundamentalmente el TNF-α. En los últimos años, no obstante, se ha incrementado notablemente nuestro conocimiento sobre las fuentes intracelulares de radicales libres y citoquinas y, en particular, del importante papel de la RI, de los AGNE y de la inflamación del tejido adiposo y hepático, lo que ha llevado a revisar el modelo patogénico original. En este sentido, ha aparecido un tercer candidato potencial a «segundo impacto» patogénico: el estrés del retículo endoplásmico (RE), y también se ha puesto de manifiesto que la apoptosis es un fenómeno frecuente en la EHNA70.
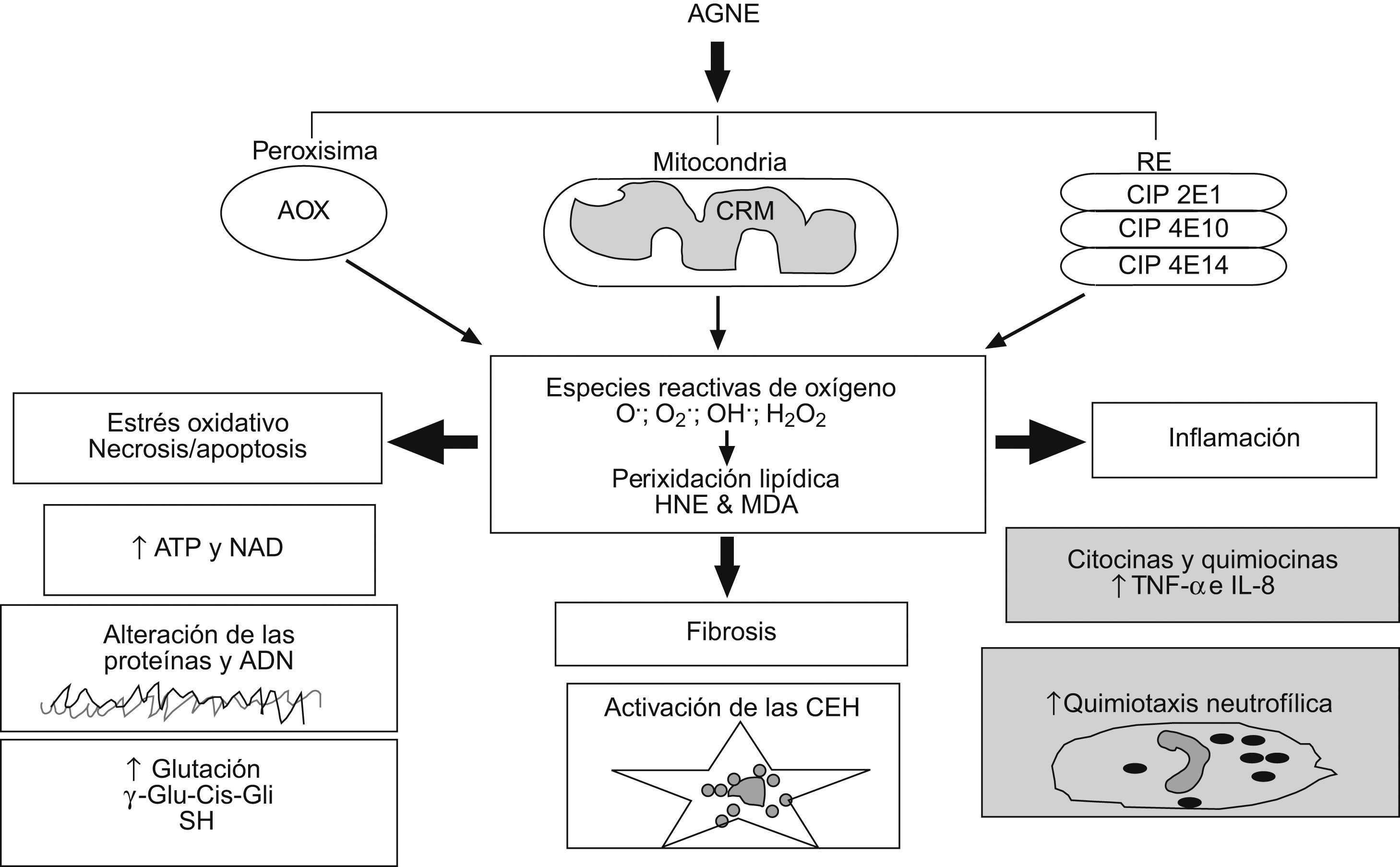
La peroxidación lipídica mediada por especies reactivas de oxígeno (ERO) es uno de los más firmes candidatos como «segundo impacto» en la patogenia de la EHNA, ya que explicaría todas las lesiones histológicas características de esta enfermedad hepática (fig. 3)71. La peroxidación de las membranas plasmática y mitocondrial puede producir directamente la muerte celular por necrosis o apoptosis y las megamitocondrias, respectivamente. También las ERO inducen la expresión hepatocelular del ligando de Fas, que puede activar la apoptosis en los hepatocitos Fas+ que se han descrito en pacientes con EHNA70. Los aldéhidos 4-hidroxinonenal y malondialdéhido, productos finales de la peroxidación lipídica, pueden formar aductos proteicos y actuar como antígenos e iniciar una respuesta inmune intrahepática72, pueden unirse a citoqueratinas hepatocelulares para formar hialina de Mallory y pueden estimular la quimiotaxis de los neutrófilos73. El estrés oxidativo puede también activar la cinasa β del inhibidor κ que, a su vez, activa el factor nuclear κβ, con el consiguiente incremento de la transcripción de genes implicados en inflamación y muerte celular74, como se ha visto en pacientes con EHNA. Además, se han encontrado marcadores de estrés oxidativo (p. ej.: proteínas nitradas en tirosina) en modelos animales y en pacientes con EHNA75,76 que relacionan la magnitud del estrés oxidativo con la gravedad de la enfermedad hepática77.
 conduce a la formación de aniones superóxido y peróxido de hidrógeno. La betaoxidación peroxisomal, iniciada por la enzima acil-CoA oxidasa, produce peróxido de hidrógeno, y la omegaoxidación en el retículo endoplásmico, catalizada por los citocromos, da lugar a diferentes especies reactivas de oxígeno. Estas y los productos finales de la peroxidación lipídica (4-hidroxinonenal y malondialdéhido) pueden provocar necrosis/apoptosis, inflamación y fibrosis. CEH: células estrelladas hepáticas; CRM: cadena respiratoria mitocondrial.")
Mecanismos del estrés oxidativo inducido por los ácidos grasos. El acúmulo de ácidos grasos no esterificados en el citosol incrementa la oxidación de éstos dentro de la célula. En las mitocondrias, la disfunción de la cadena respiratoria (CRM) conduce a la formación de aniones superóxido y peróxido de hidrógeno. La betaoxidación peroxisomal, iniciada por la enzima acil-CoA oxidasa, produce peróxido de hidrógeno, y la omegaoxidación en el retículo endoplásmico, catalizada por los citocromos, da lugar a diferentes especies reactivas de oxígeno. Estas y los productos finales de la peroxidación lipídica (4-hidroxinonenal y malondialdéhido) pueden provocar necrosis/apoptosis, inflamación y fibrosis. CEH: células estrelladas hepáticas; CRM: cadena respiratoria mitocondrial.
Clásicamente se ha considerado que las células inflamatorias son la fuente fundamental de ERO en la EHNA, pero recientemente se ha observado que los hepatocitos pueden ser una fuente importante de ERO como consecuencia de la oxidación de un exceso de AGNE en las células hepáticas78. Tanto los AGNE como sus metabolitos son ligandos del PPAR-α que, a su vez, activa la transcripción de los genes de las enzimas que participan en la oxidación mitocondrial, peroxisomal y microsomal de los AGNE, y genera ERO por al menos 3 vías diferentes que contribuyen al estrés oxidativo (fig. 3)79. Curiosamente, los ratones sin genes PPARα knockout homocigotos) son más susceptibles a una dieta deficiente en metionina y colina que los ratones normales, quizás debido a la masiva acumulación hepática de lípidos que ocurre en estos ratones80. Por otro lado, se ha descrito mayor actividad del citocromo 2E1 en el hígado de pacientes obesos con EHNA que en el de los pacientes obesos sin EHNA, lo que indica que la generación de ERO secundaria al aumento de actividad de la betaoxidación peroxisomal de los AGNE podría ser relevante en la progresión de esteatosis a esteatohepatitis75.
La mayoría de los datos provenientes de modelos animales y de pacientes con EHGNA indica, no obstante, que las mitocondrias son la fuente intracelular más importante de ERO81. Los ratones knockout homocigotos para la enzima acil-CoA oxidasa, la enzima inicial de la betaoxidación peroxisomal, desarrollan una EHNA importante, presumiblemente como consecuencia de un marcado incremento compensatorio de la betaoxidación mitocondrial y microsomal de los AGNE82. Además, se ha demostrado que las mitocondrias aisladas de hígados de ratones ob/ob generan más peróxido de hidrógeno y anión superóxido que las mitocondrias de los ratones normales83. Por otro lado, existe una evidencia creciente de que la disfunción mitocondrial que se acompaña de una generación excesiva de ERO es un trastorno muy frecuente en pacientes con EHNA84,85.
Las citoquinas son claros candidatos como mediadores en la progresión de una esteatosis simple a esteatohepatitis («segundo impacto») por varias razones. Primera, las citoquinas son capaces de reproducir todas las características histológicas clásicas de la EHNA, incluyendo necrosis/apoptosis hepatocelular (TNF-α/transforming growth factor [TGF-β]), quimiotaxis de los neutrófilos (IL-8), activación de las células estrelladas hepáticas (CEH) (TNF-α/TGF-β) e hialina de Mallory (TGF-β)86. Segundo, las citoquinas proinflamatorias (TNF-α, IL-6 e IL-1β) desempeñan un importante papel en la patogenia de la RI sistémica y hepática que tienen los pacientes con EHNA87. Tercera, las concentraciones séricas e intrahepáticas de TNF-α y de sus receptores están aumentadas en pacientes con EHGNA88,89, aunque sin discriminar claramente esteatosis de esteatohepatitis, lo que cuestiona el papel de esta citoquina en la inflamación hepática. El hecho de que los ratones knockout para TNF-α y para el receptor tipo i de TNF-α desarrollen una esteatohepatitis90,91 demuestra que esta citoquina, al menos en ratones, no es un mediador esencial en esta enfermedad. Y cuarta, el TNF-α podría ser el inductor de apoptosis en los hepatocitos de los pacientes con EHNA, ya que se ha demostrado que esta citoquina induce muerte celular programada en los hepatocitos en condiciones de estrés oxidativo92 y que éste aumenta la sensibilidad de las células hepáticas a los efectos mitocondriales del TNF-α93. Se sabe que los AGNE pueden activar directamente la vía cinasa β del inhibidor κ/factor nuclear κβ en los hepatocitos, con el consiguiente incremento en la expresión hepatocelular de TNF-α94, lo que indica que los hepatocitos pueden ser una fuente importante de esta citoquina en la EHGNA. Por otro lado, el hecho de que las células de Kupffer de los ratones con EHNA estén activadas indica que puedan ser ellas la fuente primordial de TNF-α en la EHNA95.
Como se comentó anteriormente, un candidato potencial en la progresión de esteatosis a esteatohepatitis es el estrés del RE. En esta organela, las proteínas sufren un proceso de plegamiento y oligomerización por parte de unas caperonas especializadas. Ya que esta función del RE es esencial para el procesamiento de las proteínas, estas organelas son extremadamente sensibles a cambios en la homeostasis celular. El aumento de la concentración de AG, la hiperinsulinemia, la hipoxia y las infecciones víricas pueden alterar la homeostasis del RE y provocar lo que se denomina respuesta al estrés del RE, que provoca la activación de un número de factores de transcripción y cinasas96. Esta activación transcripcional conduce a aumento de la síntesis lipídica, inducción de apoptosis, inflamación y disfunción mitocondrial97, típicas alteraciones de la EHNA. Se ha demostrado la existencia de estrés del RE en el hígado de ratones ob/ob y de ratones obesos alimentados con una dieta rica en grasas98, en cambio, no hay evidencia directa de la participación del estrés del RE en la patogenia de la EHNA, aunque sí hay datos que implican a este trastorno en la hepatopatía alcohólica99.
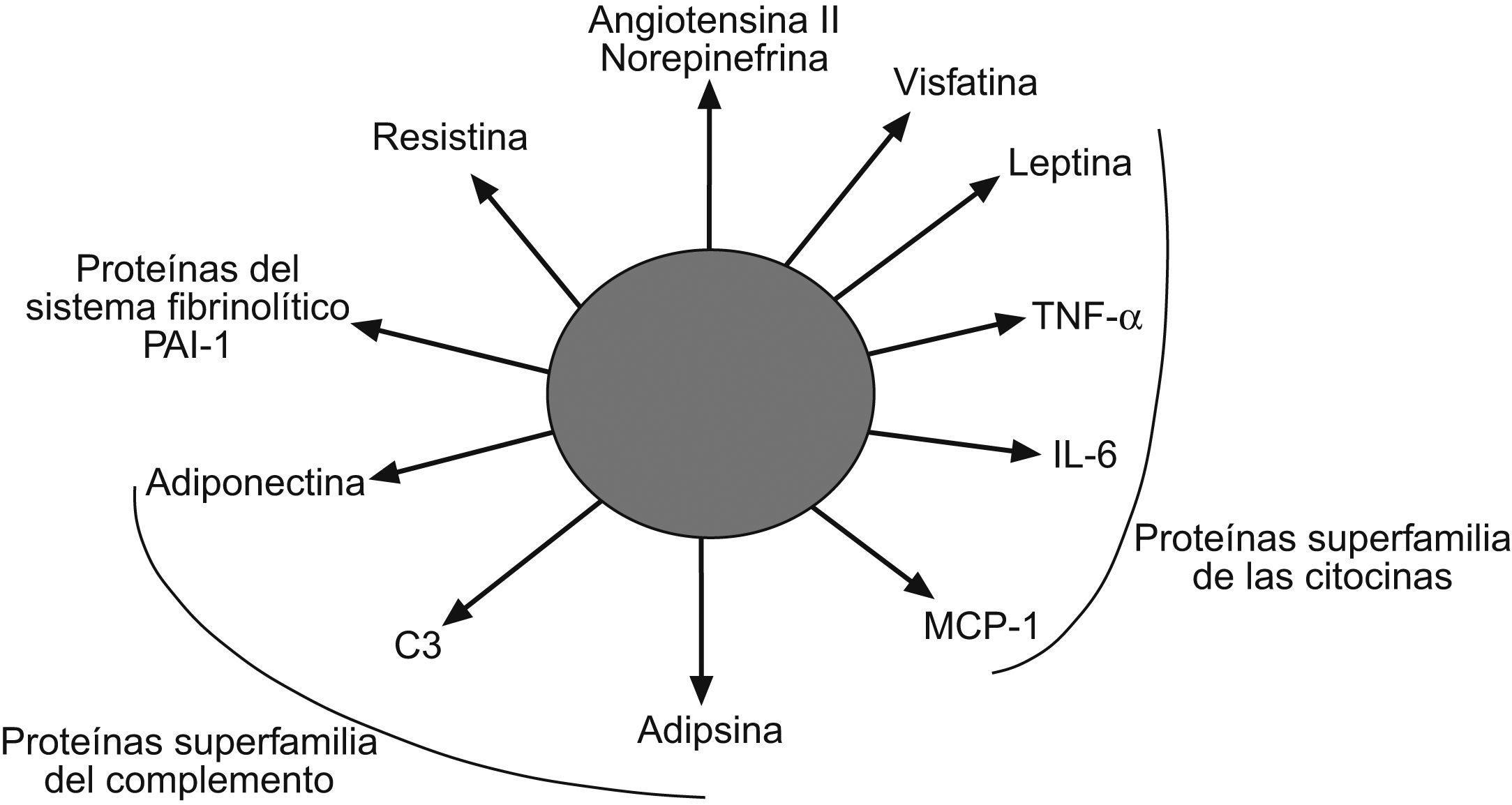
El concepto de que el tejido adiposo es un mero y pasivo almacén de triglicéridos ha cambiado considerablemente en los últimos años. Al tejido adiposo se lo considera actualmente como un tejido endocrino complejo y activo que secreta numerosos mediadores que desempeñan importantes funciones reguladoras de la biología vascular y metabólica100. Las células adiposas, que incluyen a los adipocitos, los preadipocitos y los macrófagos, son capaces de secretar múltiples moléculas bioactivas que se denominan colectivamente adipoquinas (fig. 4). Entre éstas se incluyen el TNF-α, la adiponectina, la leptina, la resistina, la IL-6, la angiotensina ii, la norepinefrina, la adipsina, la visfatina y algunas más. Ciertas adipoquinas actúan predominantemente de una manera autocrina o paracrina, mientras que otras se secretan a la circulación sistémica y actúan como moléculas señalizadoras en tejidos distantes, como el hígado, el músculo y el endotelio; es decir, pueden actuar como verdaderas hormonas100. Evidencias experimentales indican que la leptina y la adiponectina podrían desempeñar un papel importante en la patogenia de la EHGNA. Se ha comprobado que la administración de leptina a pacientes y ratones con lipodistrofias congenitas que cursan con una práctica ausencia de leptina revierte la EHGNA101,102. En cambio, en la EHGNA asociada a obesidad y RI, las concentraciones séricas de leptina están elevadas de manera proporcional al grado de esteatosis hepática103,104. En estos pacientes parece existir un estado de «resistencia a la leptina» que acompaña a la RI y hace que el hígado se vuelva refractario a los beneficiosos efectos antiesteatósicos de la leptina103. Esta adipoquina podría crontribuir a la inflamación hepática debido a su capacidad para estimular la secreción hepatocelular de una citoquina proinflamatoria de tipo Th1 denominada osteopontina. En apoyo a esta hipótesis, los ratones knockout para osteopontina no desarrollan inflamación hepática ni fibrosis cuando se les induce una esteatohepatitis con una dieta rica en grasas105.

Existen evidencias clínicas y experimentales que indican que la adiponectina puede desempeñar un importante papel en la patogenia de la EHGNA. Así, se ha visto que las concentraciones séricas de adiponectina son inversamente propocionales al grado de esteatosis hepática89,106, y en un estudio se observó que las concentraciones circulantes de adiponectina eran menores en los casos de EHNA que en aquéllos con esteatosis simple89. En la misma línea, otro estudio demostró que el nivel de expresión intrahepática de adiponectina y de su receptor tipo ii era también menor en la EHNA que en la esteatosis107. La adiponectina es una hormona antiesteatósica que promueve la betaoxidación mitocondrial de los AGNE y ejerce este efecto a través de la activación del PPAR-α y de la AMPK108. La adiponectina también posee un efecto antiinflamatorio que probablemente se deba a su capacidad para inhibir la síntesis y la secreción de TNF-α por parte de los macrófagos que infiltran el tejido adiposo en la obesidad109. Curiosamente, estos 2 factores se regulan mutuamente su actividad biológica. El TNF-α inhibe la síntesis y la actividad de la adiponectina y ésta inhibe la síntesis y la actividad del TNF-α. La importancia de la adiponectina se vió reforzada por la demostración de que su administración a ratones ob/ob revertía la esteatosis y la esteatohepatitis110. Estudios posteriores en otros modelos animales de EHNA confirmaron que la relación TNF-α alto/adiponectina baja promovía la EHNA en ratones111. Un trabajo del grupo del Dr. Farell indica que un mecanismo similar puede ser igualmente importante en humanos89, ya que al estudiar una serie de 80 pacientes con EHNA encontró que una característica común era la presencia combinada de concentraciones séricas elevadas de TNF-α y bajas de adiponectina, así como que la hipoadiponectinemia se asociaba significativamente a grados más altos de inflamación hepática. Sobre la base de estos datos clínicos y experimentales, tanto en humanos como en ratones, el desequilibrio entre TNF-α y adiponectina parece desempeñar un papel crucial en la progresión de la EHGNA.
Cualquiera de los mecanismos necroinflamatorios descritos hasta ahora podría activar a las CEH, las principales implicadas en el proceso de fibrogénesis hepática. En este sentido, se ha indicado que la apoptosis hepatocitaria, una alteración frecuente en la EHNA70, podría activar la maquinaria fibrogénica de las CEH al inducir la secreción de TGF-β, tanto por parte de las células de Kupffer como por las mismas CEH que fagocitan los hepatocitos apotóticos112,113. También se han descrito mecanismos no necroinflamatorios, más bien relacionados con la obesidad y la RI, en la patogenia de la fibrosis hepática asociada a la EHGNA. La leptina es esencial para que se produzca fibrosis hepática en modelos animales de EHNA114 pero, en pacientes con EHGNA, las concentraciones séricas de leptina están elevadas aunque sin una clara asociación con el estadío fibrótico de la enfermedad hepática104. También se ha visto que la angiotensina ii y la norepinefrina pueden activar la fibrogénesis in vitro y en ratones ob/ob deficientes en leptina115,116. La insulina y la glucosa son capaces de estimular la síntesis del factor de crecimiento del tejido conectivo en las CEH in vitro117. Además, la expresión intrahepática de este factor de crecimiento se correlaciona positivamente con el estadio fibrótico de pacientes con EHNA117, lo que indica que la hiperinsulinemia y la hiperglucemia podrían jugar un importante papel en la progresión de la fibrosis hepática en estos pacientes.
La obesidad contribuye a la progresión de enfermedades hepáticas de diversas etiologíasDiferentes estudios han demostrado de manera fehaciente que la obesidad desempeña también un papel importante en la progresión de enfermedades hepáticas de etiología conocida, de las que la hepatitis crónica por VHC es el ejemplo mejor definido118.
La esteatosis asociada a la infección crónica por el genotipo 3 del VHC se produce como consecuencia de alteraciones de la homeostasis lipídica hepatocelular inducidas por el virus119, mientras que se considera que la esteatosis en la infección crónica por genotipos del VHC diferentes al 3 se debe fundamentalmente a factores metabólicos del huésped120, como la obesidad y la diabetes, aunque exacerbados por la infección vírica121. Se ha demostrado que la obesidad y las alteraciones metabólicas que conlleva, como la RI, la diabetes y la esteatosis, contribuyen a la progresión de la fibrosis hepática en los pacientes con infección crónica por el VHC y a una menor tasa de respuesta al tratamiento antiviral122–128, por lo que actualmente se está proponiendo la implementación de estrategias terapéuticas encaminadas a la normalización de las alteraciones metabólicas de los pacientes con hepatitis crónica C antes y durante el tratamiento antiviral.
La obesidad también tiene un efecto deletéreo sobre otras enfermedades hepáticas como la hemocromatosis hereditaria y la hepatopatía alcohólica, y contribuye a un mayor riesgo de esteatosis en estas entidades4,14.
ConclusionesAunque se sabe desde hace más de 40 años que la obesidad se asocia a alteraciones hepáticas, no ha sido hasta hace poco más de 10 años que realmente se ha reconocido a la obesidad como un factor etiológico de la EHGNA y como un factor de riesgo de progresión a cirrosis, tanto en la EHGNA como en otras hepatopatías crónicas de etiología vírica o por abuso de alcohol. En los últimos años se ha producido un enorme avance en el conocimiento de los mecanismos patogénicos de las alteraciones hepáticas asociadas a la obesidad. El descubrimiento de que el tejido adiposo, y en especial el que se acumula en el abdomen, es un tejido sometido a un estado de inflamación crónica capaz de secretar adipoquinas ha permitido establecer un nexo de unión entre las alteraciones metabólicas que conducen al acúmulo de triglicéridos y a la inflamación hepática, lo que refuerza el papel de la lipotoxicidad hepatocelular en la patogenia de la EHGNA (fig. 5). Por otro lado, aunque el genotipo 3 del VHC induce esteatosis, actualmente se considera que la obesidad y sus alteraciones metabólicas asociadas, como la RI, están implicadas en la progresión de la enfermedad hepática mediada por el VHC así como en la de otras hepatopatías crónicas de diversas etiologías. Cuanto más amplio y detallado sea nuestro conocimiento de las alteraciones moleculares implicadas en la patogenia de los trastornos hepáticos relacionados con la obesidad, mayor será la probabilidad de identificar potenciales dianas que puedan ser útiles para el diseño de nuevas estrategias para su prevención y su tratamiento. En cualquier caso, independientemente de que se disponga de un tratamiento farmacológico eficaz, el tratamiento de los pacientes obesos con una enfermedad hepática asociada debería estar enfocado por un equipo multidisciplinario (en el que se incluyan profesionales expertos en nutrición, psicólogos y fisioterapeutas) con el fin de motivar a estos pacientes para que adopten un estilo de vida más saludable, disminuyan el exceso de nutrientes hipercalóricos y aumenten el ejercicio físico regular y aeróbico. Estas medidas deberían también promoverse en la sociedad por parte de las autoridades sanitarias con el objetivo de frenar la epidemia del siglo xxi: la obesidad.

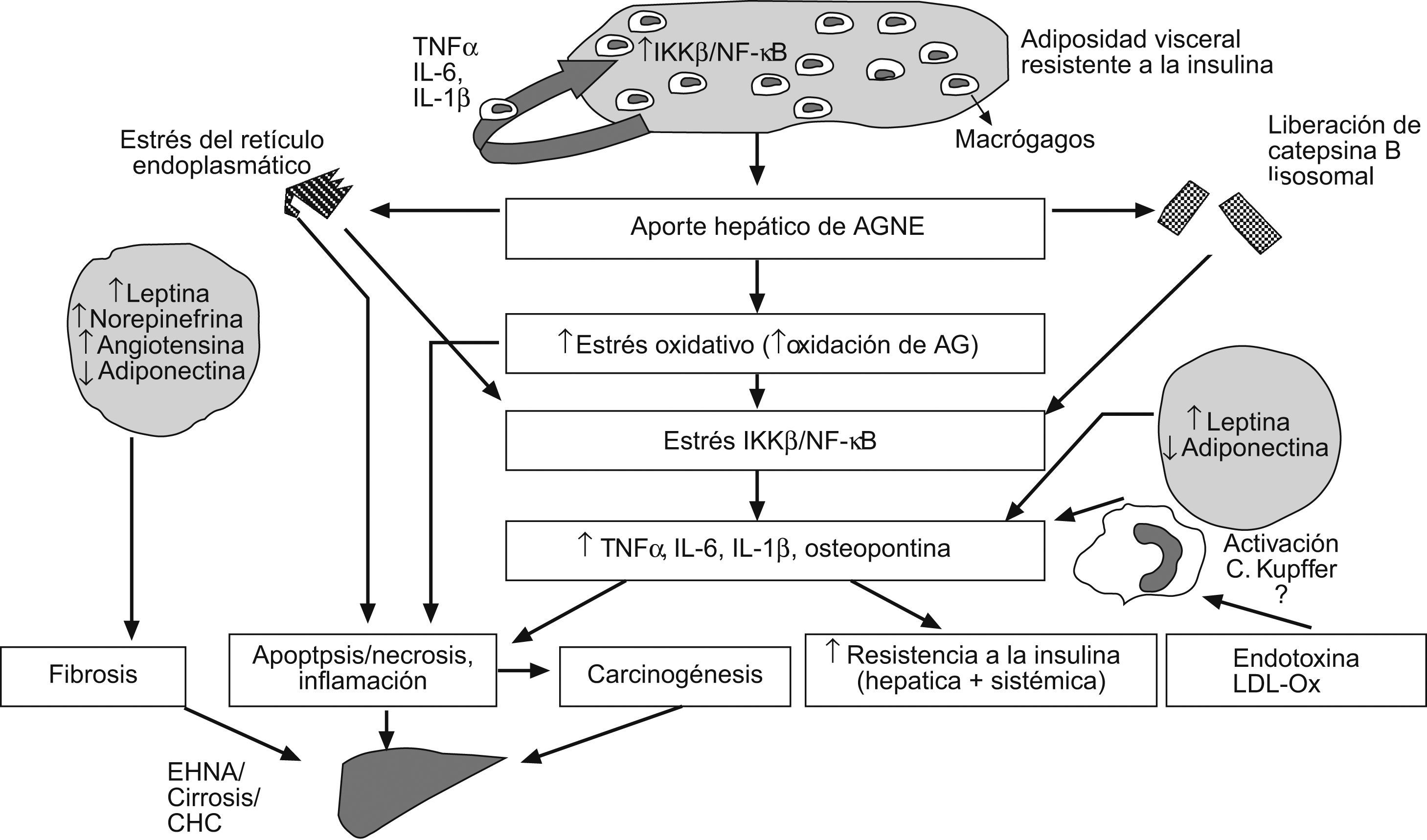
Esquema integrado del modelo patogénico de la enfermedad hepática por depósito de grasa no relacionada con el abuso de alcohol. El incremento del aporte hepático de ácidos grasos no esterificados como consecuencia de la lipólisis mantenida del tejido adiposo visceral induce estrés oxidativo, liberación de catepsina B lisosomal y estrés del retículo endoplásmico en el hepatocito. Esta situación inicia la lipoperoxidación de las membranas y activa la cascada de señalización mediada por el factor nuclear κβ, lo que se traduce en la transcripción de citoquinas, como el TNF-α y la IL-6, que son determinantes en los mecanismos de resistencia a la insulina, la apoptosis, la inflamación y la fibrosis. Tanto los macrófagos del tejido adiposo como las células de Kupffer del hígado pueden también producir TNF-α tras la estimulación por toxinas bacterianas y otras sustancias procedentes del intestino. Finalmente, desde el tejido adiposo visceral el aumento de la secreción de leptina y otras adipoquinas, así como la disminución de adiponectina, contribuyen al daño hepático característico de la enfermedad hepática por depósito de grasa no relacionada con el abuso de alcohol.
Los autores declaran no tener ningún conflicto de intereses.
FinanciaciónEl CIBERehd está financiado por el Instituto de Salud Carlos III.

