




La poliposis adenomatosa familiar (PAF) se caracteriza principalmente por el desarrollo de un gran número de pólipos en el tracto gastrointestinal y por un mayor riesgo de desarrollo de adenocarcinomas. A continuación presentamos el caso de una paciente diagnosticada de PAF y metástasis hepáticas, cuyo examen histológico reveló que eran secundarias a un tumor neuroendocrino. En la revisión bibliográfica sólo se han descrito hasta el momento 3 casos en los cuales ambas entidades coexisten. Actualmente no hay ninguna base genética que explique la coexistencia de estas 2 enfermedades cuyas prevalencias son muy bajas.
Familial adenomatous polyposis (FAP) is mainly characterized by the development of a large number of polyps in the gastrointestinal tract and by the risk of developing adenocarcinomas. We present the case of a woman diagnosed with FAP and liver metastases. Histological analysis revealed both diseases to be secondary to a neuroendocrine tumor. To date, only three cases showing the simultaneous occurrence of these two entities have been published. Currently, there is no genetic basis to explain the coexistence of these two diseases, both of which have a very low prevalence.
La polipomatosis adenomatosa familiar (PAF) es un síndrome hereditario caracterizado principalmente por la presencia de múltiples adenomas colónicos, en número superior a 100. Se trata de una entidad poco frecuente, cuya prevalencia se estima en 1 por cada 5.000 a 7.5001.
Este síndrome está causado por una mutación germinal con herencia autosómica dominante, en el gen de la poliposis adenomatosa coli (APC), localizado en el cromosoma 5q21-q222. En un tercio de los pacientes no existen antecedentes familiares de la enfermedad, tratándose por tanto de mutaciones de novo. Por otra parte, se ha identificado una forma atenuada de PAF caracterizada por un menor número de adenomas colorrectales, entre 15-100, como consecuencia de mutaciones bilalélicas, en la línea germinal del gen MYH, localizado en el cromosoma 1p34.3-1p32.1, con patrón de herencia autosómica recesiva.
La proteína APC es un regulador multifactorial de la homeostasia de las células epiteliales colónicas y participa en procesos de proliferación, migración, diferenciación, apoptosis celular y segregación cromosómica3.
Por otra parte, los tumores neuroendocrinos se originan en las células del sistema neuroendocrino del tracto gastrointestinal4. La incidencia se estima en 1-2 casos por cada 100.000 personas al año. La localización más frecuente es el íleon, seguida del recto, aunque en los últimos años ha aumentado la incidencia de lesiones neuroendocrinas en estómago. Generalmente son asintomáticos, tienen un crecimiento lento, y aunque potencialmente tiene la capacidad de producir metástasis, esta complicación es infrecuente y se asocia sobre todo con aquellos con un tamaño mayor de 1cm5,6.
Los tumores neuroendocrinos pueden presentarse de forma esporádica o asociados a la neurofibromatosis tipo I, el síndrome de Zollinger-Ellison o el síndrome de neoplasia endocrina múltiple 1 (síndrome MEN I). Sin embargo, no se ha establecido ninguna asociación con la PAF.
Caso clínicoMujer de 24 años, que acudió a urgencias por dolor abdominal. Durante su estancia en dicho servicio se solicitó una analítica de sangre. Las únicas alteraciones analíticas observadas eran un aumento de la amilasa 931 U/l (22-80) e hipercalcemia de 14,59mg/dl (8,5-10,6), motivo por el cual la paciente ingresó con el diagnóstico de pancreatitis aguda e hipercalcemia. Como antecedentes personales, la paciente había sido intervenida en la infancia por tumores desmoides recidivantes sobre la escápula izquierda. En la exploración física destacaba una hepatomegalia gigante dolorosa. La ecografía abdominal mostró la presencia de hepatomegalia causada por la existencia de múltiples lesiones sólidas indicativas de metástasis. La vesícula contenía microlitiasis y la vía biliar no estaba dilatada. Aunque inicialmente se relacionó la etiología de la pancreatitis a la hipercalcemia, el hallazgo de microlitiasis en la ecografía abdominal, a pesar de la ausencia de alteraciones de las enzimas hepáticas, no permite descartad la etiología biliar como posible desencadenante de la pancreatitis.
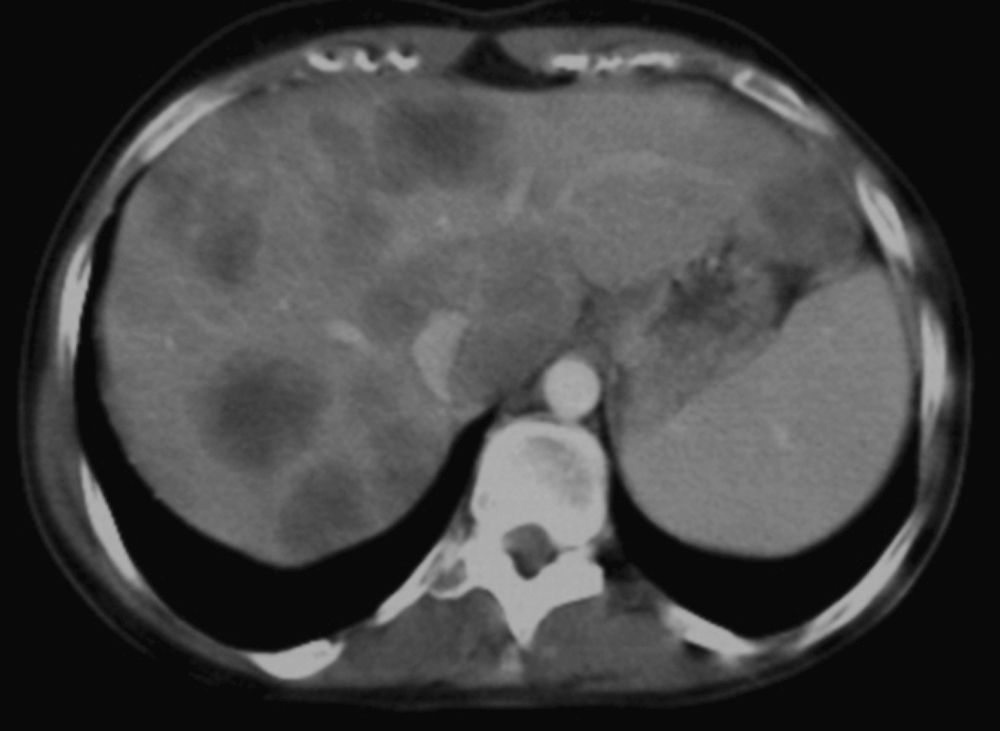
La TAC realizada posteriormente confirmaba la presencia de múltiples lesiones focales hepáticas con realce en anillo compatibles con metástasis (fig. 1) y un engrosamiento de la pared de rectosigma. Ante los hallazgos de la TAC se solicitó una colonoscopia en la que se describía una afectación de toda la mucosa de recto y colon, tapizada por múltiples pólipos de tamaño variable, entre 5mm y 4cm, algunos de ellos con amplia base de implantación, irregulares y ulcerados. Por encima del margen anal presentaba una tumoración sésil, que ocupaba un cuarto de la circunferencia del recto, con ulceración central, superficie irregular e indurada. Las biopsias de esta última lesión confirmaron la presencia de un adenocarcinoma bien diferenciado infiltrante. El resto de los pólipos biopsiados fueron informados como adenomas tubulares con displasia de bajo grado, algunos de ellos con displasia focal de alto grado. Ante la sospecha de una PAF por la presencia de numerosos pólipos colónicos y el antecedente de tumor desmoide recidivante en la adolescencia, se realizó una gastroscopia que mostraba múltiples pólipos gástricos sésiles de 2-4mm que se biopsiaron, siendo el resultado del estudio anatomopatológico de pólipos de glándulas fúndicas. En la segunda porción duodenal se observaron múltiples pólipos sésiles de menos de 2-3mm, con el diagnóstico anatomopatológico de adenomas tubulares con displasia de bajo grado.

Los marcadores tumorales mostraban un leve aumento del CA 125: 54,80 UI/ml (0-35) y CEA 6,60 ng/ml (0-5). Los otros marcadores solicitados, CA 15.3 y CA 19.9, estaban en los límites de la normalidad: CA 15.3: 7,20 UI/ml (0-31), CA 19.9: < 2 UI/Ml (0-37).
La resonancia magnética cerebral, ecografía tiroidea, radiografía de cráneo y mandíbula y la valoración oftalmológica realizadas no mostraron anomalías. En la ortopantomografía se observó la presencia de dientes supernumerarios.
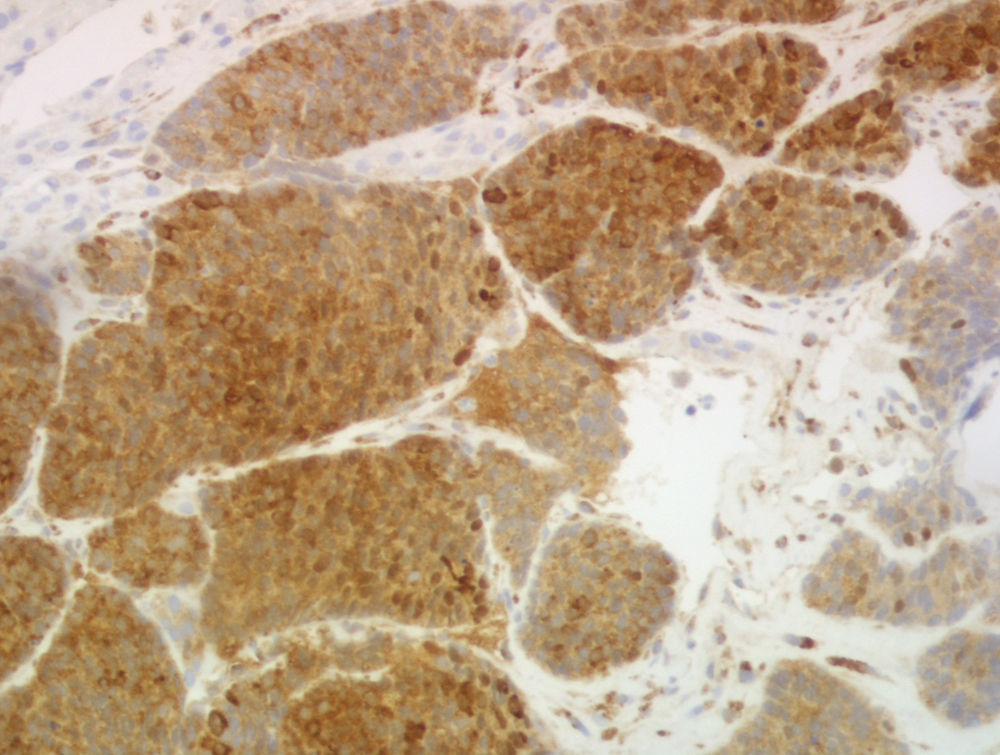
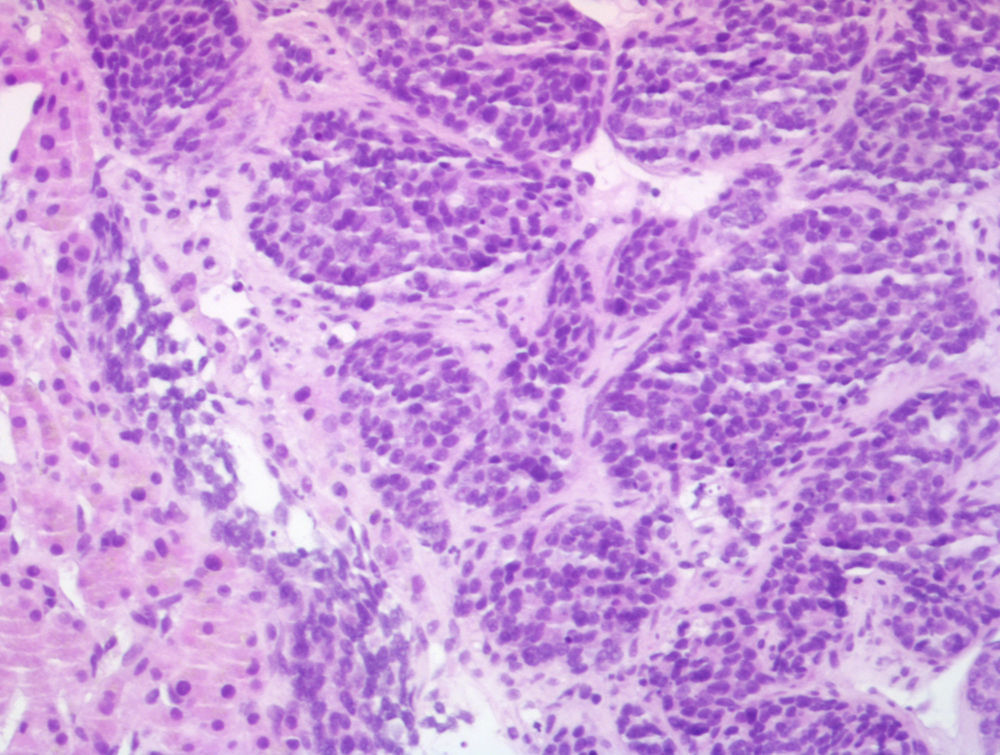
Para completar el estudio se realizó una biopsia de las lesiones hepáticas, en la que se objetivó infiltración por carcinoma neuroendocrino de alto grado, con positividad para cromogranina, enolasa y EMA y negatividad para CK7 y CK20, con un índice mitótico de 10 mitosis/10 campos de gran aumento y un índice de proliferación (Ki-67) mayor del 30% (figs. 2 y 3).

.")
Para localizar el origen de las metástasis hepáticas se solicitó un octreoscan en el cual se confirmó la presencia de múltiples focos captantes compatibles con metástasis hepáticas sin poder evaluar el páncreas por el aumento de tamaño del hígado ni identificar otro foco captante. La resonancia de páncreas no mostró alteraciones. Las determinaciones de 5-hidroxiindolacético en orina y serotonina en suero fueron normales. La cromogranina A mostraba un leve aumento: 5,1 nmol/l (normal < 4; dudoso 4-10).
Se consultó el caso con el servicio de oncología médica. Con el diagnóstico de metástasis hepáticas de carcinoma neuroendocrino mal diferenciado (Ki-67 > 30%) y la presencia de un adenocarcinoma rectal, se decidió tratar el primer tumor, de peor pronóstico, iniciando quimioterapia con esquema cisplatino y etopósido, además de la administración de ácido zolendrónico para el control de la hipercalcemia. Las metástasis hepáticas mejoraron con el tratamiento, sin embargo el adenocarcinoma de recto sufrió un empeoramiento progresivo, con sangrado y dolor de difícil control. Se solicitó valoración por el servicio de cirugía, que desestimó el tratamiento quirúrgico. Finalmente la paciente ingresó por cuadro clínico de oclusión intestinal y rectorragia incoercible a pesar de las medidas locales aplicadas, falleciendo 9 meses después del diagnóstico.
La paciente había sido remitida a la unidad de consejo genético en cáncer. Ante la inexistencia de antecedentes familiares se sospechó que muy probablemente se trataba de una mutación de novo. El estudio genético confirmó una mutación en el gen APC: cambio de base C>T en la posición 4348 del ADNc en el fragmento H del exón 15 (R1450X) en heterocigosis. Dicho cambio provoca la aparición de un codón de parada temprano y origina una proteína truncada. En la actualidad está pendiente el resultado del estudio directo en un familiar de primer grado, mientras que los otros 2 familiares han rechazado el estudio.
DiscusiónLos pacientes afectados por la PAF también tienen un mayor riesgo de neoplasias extracolónicas como el desarrollo de carcinoma en la papila duodenal, cáncer de tiroides folicular o papilar, hepatoblastoma, carcinomas gástricos o tumores en el sistema nervioso central7. Distintos estudios han correlacionado las diferentes mutaciones en el gen APC con determinados fenotipos. Aquellas mutaciones entre los codones 1250 a 1464 se asocian con la forma clásica de la PAF, es decir, con la presencia de cientos a miles de adenomas colónicos8. Las localizadas entre los codones 1445 y 1578 se asocian con el desarrollo de tumores desmoides en algunos casos9,10. Otras localizadas por debajo del codón 1051 se han asociado a la presencia de lesiones con displasia avanzada periampulares11, y aquellas mutaciones localizadas en la parte central del gen APC (entre los codones 279 a 1309) se correlacionan con el desarrollo de poliposis duodenal12. Sin embargo, hasta el momento no se ha descrito un aumento del riesgo de tumores neuroendocrinos entre los pacientes con PAF.
En la revisión bibliográfica realizada tan sólo se han descrito 3 casos de pacientes en los que se observase la presencia de tumores neuroendocrinos en pacientes afectados por PAF. En uno de los casos descritos se diagnosticó la presencia de un tumor carcinoide localizado en el cuerpo gástrico13. En los otros 2 casos se localizaban en el duodeno y la región periampular14,15.
Como hemos mencionado anteriormente la probabilidad de presentar una PAF o de desarrollar un tumor neuroendocrino es baja, por tanto la presencia de forma conjunta de ambas entidades plantea la posibilidad de que no se trate de una mera coincidencia, sino que exista una vía común, actualmente no identificada, que predisponga al desarrollo de tumores neuroendocrinos en los pacientes afectados de PAF.
Una de las posibles hipótesis planteadas, como mecanismo común de ambas entidades, sería la alteración en la regulación de la vía de señalización de la Wnt/ß-catenina, ya que se han descrito mutaciones en la misma tanto en casos de tumores neuroendocrinos como en algunos casos de pacientes con PAF16. Otros autores por el contrario, mostraron que ni las mutaciones en el exón 3 del gen de la β-catenina, ni las del exón 15 del gen APC contribuían a la activación de la vía Wnt/β-catenina/APC en los tumores neuroendocrinos gastrointestinales17. Sin embargo, en este caso no se estudiaron el resto de exones en estos genes ni tampoco las zonas reguladoras de éstos, por lo que no se puede descartar la hipótesis de una alteración en la vía de señalización de la Wnt/ß-catenina o en otras localizaciones aún no estudiadas hasta el momento. Por tanto, es evidente que se precisan más investigaciones que demuestren las posibles alteraciones que predispongan al desarrollo de tumores neuroendocrinos en los pacientes con PAF.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

