




La pancreatitis autoinmune es una enfermedad fibroinflamatoria benigna del páncreas de probable origen autoinmune, que engloba 2 tipos diferentes: tipo 1 (pancreatitis esclerosante linfoplasmocítica) y tipo 2 (pancreatitis ductal central idiopática). Es frecuente su presentación clínica como ictericia obstructiva en un paciente con una masa pancreática, por lo que debe formar parte del diagnóstico diferencial de la neoplasia pancreática. A lo largo de la historia se han descrito numerosos criterios diagnósticos, siendo los más empleados los criterios HISORT de la clínica Mayo y los del Consenso Internacional de 2011. Su principal característica en la respuesta al tratamiento esteroideo, sin necesidad de ningún tratamiento quirúrgico. Conocer esta enfermedad y hacer un correcto diagnóstico y tratamiento puede cambiar de forma radical el manejo de un paciente con ictericia obstructiva, que de otra forma sería sometido a una duodenopancreatectomía.
Autoimmune pancreatitis is a benign fibroinflammatory disease of the pancreas of probable autoimmune origin, which includes 2 different phenotypes: type 1 (lymphoplasmacytic sclerosing pancreatitis) and type 2 (idiopathic duct-centric pancreatitis). Its clinical presentation as obstructive jaundice in patients with a pancreatic mass is common and therefore it must be included in the differential diagnosis of pancreatic neoplasia. Many diagnostic criteria have been described throughout history. The most famous are the HISORT criteria of the Mayo Clinic and the international consensus criteria of 2011. One of the main features of autoimmune pancreatitis is its dramatic response to steroid therapy, without the need for surgical treatment. Knowledge of this disease can dramatically change the management of patients with obstructive jaundice, who would otherwise be subjected to a pancreaticoduodenectomy.
La pancreatitis autoinmune (PAI) es una enfermedad fibroinflamatoria benigna del páncreas de probable origen autoinmune. Desde la publicación en 1961 del primer artículo donde se menciona la pancreatitis crónica idiopática asociada a hipergammaglobulinemia, esta entidad ha recibido diferentes nombres como pancreatitis tumefactiva o pancreatitis destructiva no alcohólica1. La PAI se puede entender bien como un trastorno primario, o bien en el contexto de enfermedades relacionadas con la inmunoglobulina (Ig) G4. Su prevalencia no está bien definida, hablándose de un 15% de las pancreatitis crónicas en las series médicas y un 26% de los procesos pancreáticos benignos en las series quirúrgicas2. Su distribución geográfica es mundial, aunque es más frecuente en los países asiáticos3.
La relevancia de esta entidad radica en ser parte del diagnóstico diferencial de la neoplasia pancreática. Saber reconocerla y aplicar el tratamiento médico adecuado pueden evitar intervenciones quirúrgicas innecesarias, que con frecuencia se llevan a cabo en este tipo de pacientes.
Este artículo pretende hacer una revisión de los conocimientos actuales, todavía incompletos, sobre la PAI.
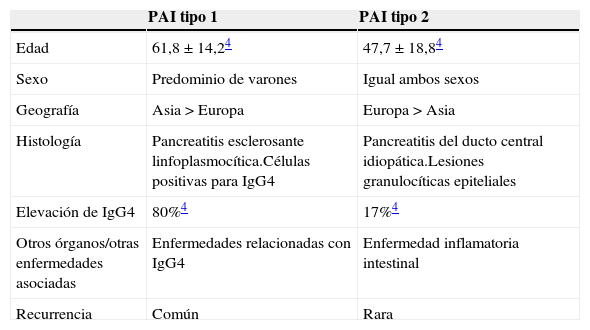
TiposDesde 2003 se han descrito 2 patrones histológicos diferentes de la PAI: tipo 1 (pancreatitis esclerosante linfoplasmocítica) y tipo 2 (pancreatitis ductal central idiopática). Actualmente, se mantiene por consenso el mismo nombre a las 2, aunque difieren entre sí en varios aspectos clínicos, epidemiológicos e histológicos4 (tabla 1).
Diferencias entre pancreatitis autoinmune tipo 1 y tipo 2
| PAI tipo 1 | PAI tipo 2 | |
|---|---|---|
| Edad | 61,8±14,24 | 47,7±18,84 |
| Sexo | Predominio de varones | Igual ambos sexos |
| Geografía | Asia>Europa | Europa>Asia |
| Histología | Pancreatitis esclerosante linfoplasmocítica.Células positivas para IgG4 | Pancreatitis del ducto central idiopática.Lesiones granulocíticas epiteliales |
| Elevación de IgG4 | 80%4 | 17%4 |
| Otros órganos/otras enfermedades asociadas | Enfermedades relacionadas con IgG4 | Enfermedad inflamatoria intestinal |
| Recurrencia | Común | Rara |
Ig: inmunoglobulina; PAI: pancreatitis autoinmune.
Muchos datos sugieren el origen autoinmune de la enfermedad: el infiltrado linfoplasmocitario en el tejido pancreático5; la respuesta a glucocorticoides6,7; los distintos tipos de anticuerpos8–10; o el tratamiento efectivo en pacientes corticorrefractarios con fármacos anti-CD2011.
La PAI tipo 1 puede entenderse como la manifestación pancreática de un trastorno multiorgánico (espectro de enfermedades IgG4). Teóricamente, esta inmunoglobulina no puede interaccionar con antígenos, formar complejos antígeno-anticuerpo ni activar la cascada de complemento. Por este motivo no es raro que no se hayan detectado antígenos específicos de IgG4 en la PAI. Las IgG4 se liberan tras la exposición prolongada a un antígeno, y todo apunta a que el mecanismo patogénico de esta entidad esté mediado por células T, que de forma secundaria activan células B, quienes liberan este tipo de Ig12.
La PAI tipo 2 en cambio, no tiene una elevación sérica de IgG4. Esto, junto al infiltrado linfoplasmocitario en la glándula pancreática, sugiere que más que tratarse de un desorden sistémico con afectación pancreática, sea una enfermedad autoinmune con el páncreas como órgano diana.
En cuanto a la susceptibilidad genética, parece haber polimorfismos en el gen del antígeno 4 de los linfocitos T citotóxicos (CTLA-4, CD152) que dan cierta predisposición a la enfermedad13.
ClínicaLa PAI puede manifestarse en la fase aguda como ictericia obstructiva o en una fase más tardía como masa pancreática, atrofia o incluso calcificaciones. El 85% de los casos presentan una masa pancreática. La presencia de esta junto con una ictericia indolora obliga a descartar como principal sospecha diagnóstica el adenocarcinoma pancreático14. La ictericia suele ser indolora, muchas veces con carácter fluctuante, secundaria a la compresión del conducto biliar por la inflamación pancreática. En ocasiones, los pacientes refieren dolor aunque suele ser de carácter leve-moderado. En la PAI tipo 2 es más frecuente la presencia de dolor abdominal y la presentación como pancreatitis aguda recurrente ocurre hasta en un tercio de los casos15.
En los casos muy evolucionados puede haber síntomas derivados de la insuficiencia exocrina (esteatorrea, déficit de vitaminas liposolubles, desnutrición) y endocrina (diabetes mellitus)16,17.
En los pacientes con PAI tipo 1 pueden darse además síntomas asociados a otras enfermedades del espectro asociado a IgG4.
DiagnósticoEl diagnóstico de la PAI requiere un alto índice de sospecha por parte del clínico, y debe plantearse ante cualquier paciente con síntomas biliopancreáticos, más aún si tiene antecedentes de otras enfermedades extrapancreáticas como tiroiditis, fibrosis retroperitoneal, colangitis autoinmune, etc.
En el proceso diagnóstico de la PAI, el diagnóstico diferencial principal se establece con el adenocarcinoma pancreático. Por este motivo, el 2,4-3,7% de los pacientes sometidos a una duodenopancreatectomía por sospecha de cáncer son finalmente diagnosticados de PAI18.
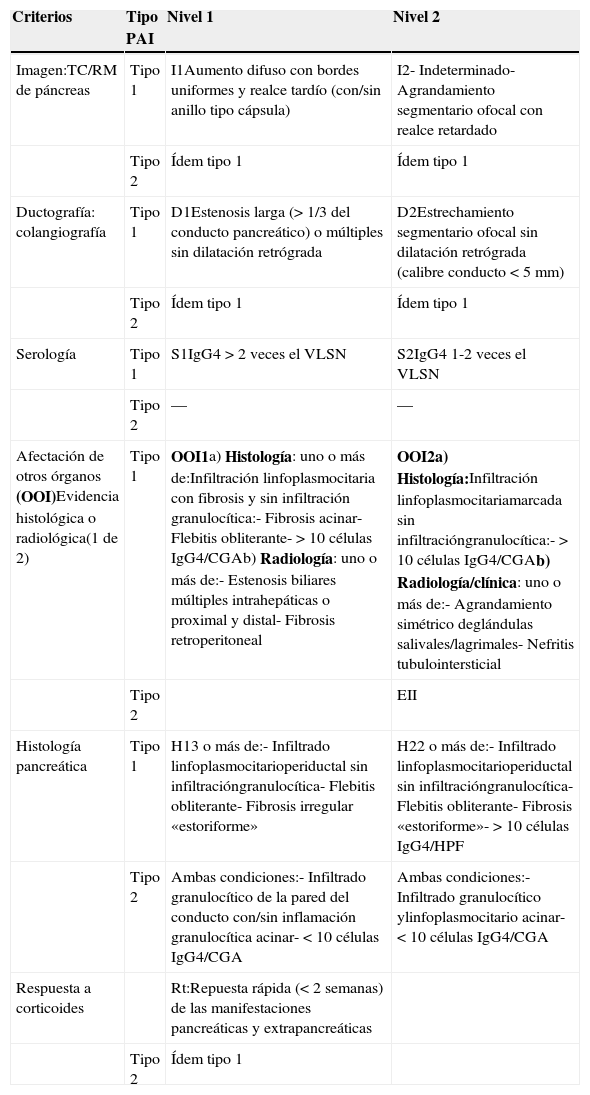
Son muchos los criterios diagnósticos reunidos a lo largo de la historia, siendo los más conocidos los criterios HISORT modificados, propuestos por la clínica Mayo en el 2009 y los del Consenso Internacional en 2011 (tabla 2)19,20. Todos ellos basan el diagnóstico en la clínica, datos de laboratorio, pruebas de imagen e histológicas.
Criterios diagnósticos del Consenso Internacional (2011)
| Criterios | Tipo PAI | Nivel 1 | Nivel 2 |
|---|---|---|---|
| Imagen:TC/RM de páncreas | Tipo 1 | I1Aumento difuso con bordes uniformes y realce tardío (con/sin anillo tipo cápsula) | I2- Indeterminado- Agrandamiento segmentario ofocal con realce retardado |
| Tipo 2 | Ídem tipo 1 | Ídem tipo 1 | |
| Ductografía: colangiografía | Tipo 1 | D1Estenosis larga (>1/3 del conducto pancreático) o múltiples sin dilatación retrógrada | D2Estrechamiento segmentario ofocal sin dilatación retrógrada (calibre conducto <5mm) |
| Tipo 2 | Ídem tipo 1 | Ídem tipo 1 | |
| Serología | Tipo 1 | S1IgG4>2 veces el VLSN | S2IgG4 1-2 veces el VLSN |
| Tipo 2 | — | — | |
| Afectación de otros órganos (OOI)Evidencia histológica o radiológica(1 de 2) | Tipo 1 | OOI1a) Histología: uno o más de:Infiltración linfoplasmocitaria con fibrosis y sin infiltración granulocítica:- Fibrosis acinar- Flebitis obliterante- >10 células IgG4/CGAb) Radiología: uno o más de:- Estenosis biliares múltiples intrahepáticas o proximal y distal- Fibrosis retroperitoneal | OOI2a) Histología:Infiltración linfoplasmocitariamarcada sin infiltracióngranulocítica:- >10 células IgG4/CGAb) Radiología/clínica: uno o más de:- Agrandamiento simétrico deglándulas salivales/lagrimales- Nefritis tubulointersticial |
| Tipo 2 | EII | ||
| Histología pancreática | Tipo 1 | H13 o más de:- Infiltrado linfoplasmocitarioperiductal sin infiltracióngranulocítica- Flebitis obliterante- Fibrosis irregular «estoriforme» | H22 o más de:- Infiltrado linfoplasmocitarioperiductal sin infiltracióngranulocítica- Flebitis obliterante- Fibrosis «estoriforme»- >10 células IgG4/HPF |
| Tipo 2 | Ambas condiciones:- Infiltrado granulocítico de la pared del conducto con/sin inflamación granulocítica acinar- <10 células IgG4/CGA | Ambas condiciones:- Infiltrado granulocítico ylinfoplasmocitario acinar- <10 células IgG4/CGA | |
| Respuesta a corticoides | Rt:Repuesta rápida (<2 semanas) de las manifestaciones pancreáticas y extrapancreáticas | ||
| Tipo 2 | Ídem tipo 1 |
Diagnóstico
Definitivo tipo 1. Cualquiera de las siguientes combinaciones:
- H1+ I (1 o 2)
- I1+cualquiera (1 o 2) no D
- I2+D2+ (≥2 de cualquier criterio nivel 1)
- Rt+I2+ (S1 o OOI 1)
- Rt.+I2+D1+(S2 o OOI 2 o H2)
Probable tipo 1. Cualquiera de las siguientes combinaciones:
- I2+(S2 o OOI 2 o H2)+Rt
Definitivo tipo 2. Cualquiera de las siguientes combinaciones:
- I (1 o 2)+H1
- I (1 o 2)+EII+H2+Rt
Probable tipo 2:
- I (1 o 2)+(H2 o EII)+Rt
CGA: campo de gran aumento; EII: enfermedad inflamatoria intestinal; VLSN: valor límite superior de la normalidad.
Fuente: Shimosegawa et al.20.
La PAI tipo 1 suele cursar con elevación serológica de IgG4, cuyo valor normal en una persona sana es el 5-6% del total de IgG (<140mg/dl). La elevación de IgG4 en la PAI tiene una sensibilidad del 53 y 76%, una especificidad del 99 y 93% y un valor predictivo positivo del 75 y 36%, si se toma como punto de corte valores de 280 y 140mg/dl, respectivamente21. El 10% de los pacientes con adenocarcinoma pancreático pueden tener niveles elevados de IgG4, pero no suelen alcanzar cifras superiores a 280mg/dl. Otras entidades en las que puede estar elevada la IgG4 son la dermatitis atópica y el eccema22. Asimismo, se ha observado en la PAI elevación de anticuerpos como los anti Protein Binding Protein (PBP), anti-anhidrasa carbónica II, anti-lactoferrina y otros como factor reumatoide, anticuerpos contra el citoplasma de los neutrófilos (p-ANCA), antimitocondriales, anti-músculo liso y anti-transglutaminasa8,9.
Por otro lado, el marcador CA19-9 puede elevarse en el 25% de las PAI, aunque no suele alcanzar valores altos, más propios de la neoplasia pancreática23.
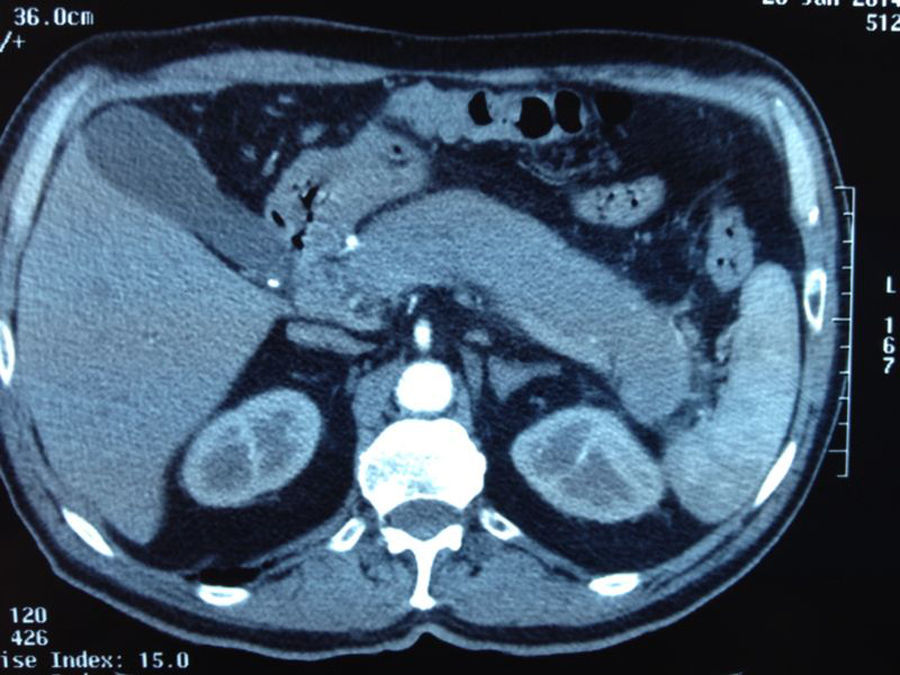
ImagenAl realizar una tomografía computarizada (TC) o resonancia magnética (RM), el aumento difuso de la glándula pancreática, con bordes uniformes y realce tardío, con/sin anillo tipo cápsula es un hallazgo muy sugestivo de PAI (fig. 1). Esta imagen, llamada «páncreas en salchicha», solo se da en el 19-27% de la PAI24. Ante este hallazgo el diagnóstico diferencial debe establecerse con el adenocarcinoma pancreático. Otro hallazgo puede ser la presencia de un engrosamiento focal. Son imágenes sugestivas de malignidad las masas de baja densidad, la dilatación del conducto pancreático con amputación brusca del mismo a nivel de la masa pancreática, así como la atrofia pancreática distal20.

En la pancreatografía es típico de la PAI la presencia de una estenosis larga del conducto pancreático principal (mayor de un tercio) o de múltiples estenosis segmentarias, sin dilatación retrógrada. Menos típica es la estenosis segmentaria o focal (calibre <5mm).
La papila mayor puede mostrar un aspecto inflamado hasta en el 25% de los casos, fruto del infiltrado linfoplasmocitario junto con células positivas para IgG425,26.
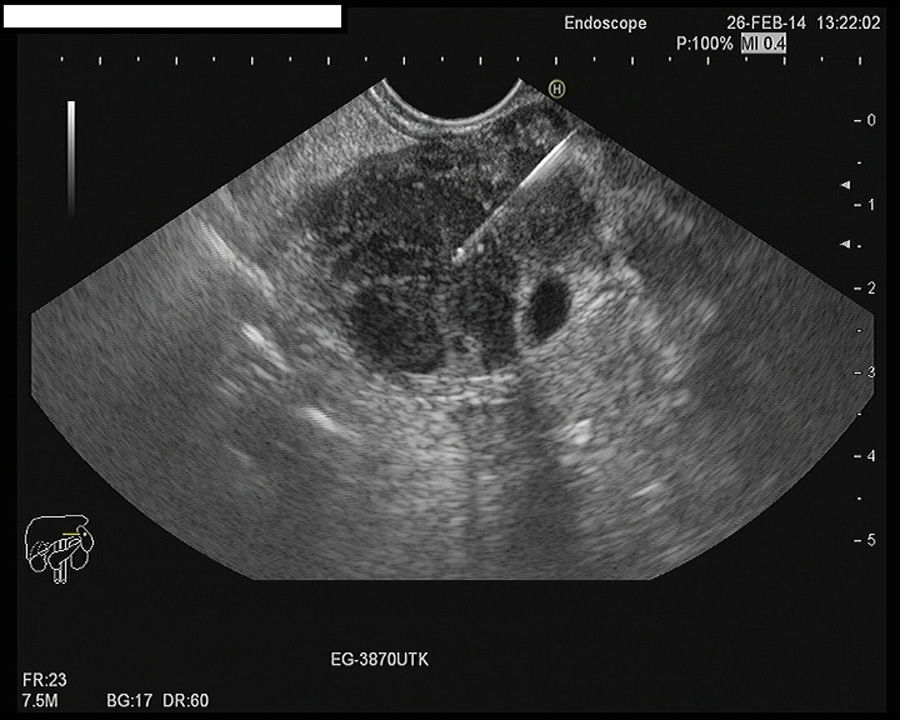
Con la ecoendoscopia o ultrasonografía endoscópica (USE) se puede visualizar un aumento difuso de la glándula, con un parénquima hipoecogénico y puntos hiperecogénicos. El hallazgo de una masa pancreática hipoecogénica puede ser difícil de distinguir del adenocarcinoma. Sin embargo, los puntos hiperecogénicos en la masa hipoecogénica y el signo del ducto penetrante (conducto pancreático no obstruido que atraviesa una masa inflamatoria) son más sugestivos de la PAI que de la neoplasia27. Otro hallazgo típico es el engrosamiento del conducto biliar principal en su porción distal, a expensas sobre todo de la capa muscular más externa28.
Anatomía patológicaLa obtención de tejido pancreático es de gran relevancia a la hora de establecer el diagnóstico de PAI. En cambio, el mejor método para obtener dicha muestra ofrece cierta controversia. En un porcentaje significativo de casos el diagnóstico se realiza tras el análisis de la pieza de pancreatectomía. Sin embargo, lo ideal sería no tener que llegar a una cirugía asociada a unas tasas de morbi-mortalidad no despreciables para diagnosticar una entidad con una excelente respuesta al tratamiento médico.
Alternativas menos invasivas son la obtención de biopsias por punción percutánea o guiada por USE. Esta última es una opción cada vez más atractiva, ya que permite hacer un examen endosonográfico del páncreas a la vez que se punciona bajo control directo, con una baja tasa de complicaciones29 (fig. 2). Las biopsias trucut permiten la obtención de un cilindro de tejido para el estudio histológico y la detección de células IgG4 por inmunohistoquímica (IHQ), aunque puede tener más complicaciones. La clínica Mayo aboga por este método ya que no se artefacta la organización tisular30. Una alternativa más cómoda y segura es la punción-aspiración con aguja fina (PAAF), con procesamiento de las muestras en bloques celulares y realización de IHQ, aunque su utilidad es más controvertida31-34. Sin embargo, hay que recalcar que la detección de células aisladas no es diagnóstica de PAI y la detección de células IgG4 en tejido no es patognomónica de la PAI tipo 135. Podría plantearse la PAAF como primer paso diagnóstico para descartar una neoplasia pancreática, ya que su precisión diagnóstica para lesiones malignas llega a ser de 94%36,37. En caso de resultado inconcluyente, el siguiente paso sería la realización de trucut biopsia mediante USE.

Las agujas ProCore® permiten la obtención de cilindros tisulares con un calibre menor que las agujas de trucut y, por tanto, con una mayor seguridad. No existen en la actualidad publicaciones respecto al uso de agujas ProCore® para el diagnóstico de la PAI, aunque existe un estudio en fase de reclutamiento (NCT01774513) cuyos resultados probablemente provoquen algunos cambios en el actual algoritmo diagnóstico de esta entidad.
Respuesta a glucocorticoidesLa respuesta rápida (<2 semanas) al tratamiento glucocorticoideo es uno de los criterios diagnósticos de la PAI. En 2008 Moon et al. estudiaron las posibles consecuencias que tendría la administración de tratamiento glucocorticoideo a pacientes con duda diagnóstica entre PAI y neoplasia tras una imagen de TC o RM no concluyente38. Observaron que tras solo 2 semanas de tratamiento ya se objetivaba una respuesta radiológica en aquellos casos de PAI. En cambio, en los que no fue así, el diagnóstico final fue siempre de neoplasia. En estos últimos, el retraso de la cirugía 2 semanas no supuso progresión de la enfermedad. En nuestra opinión, dada la rápida respuesta de la PAI al tratamiento esteroideo, los casos con presentación en forma de masa pancreática o con elevación del marcador CA19-9 deberían hacerse un control radiológico con TC o RM a las 2 o 4 semanas con el fin de excluir la neoplasia pancreática.
TratamientoEl tratamiento de elección de la PAI se basa en los corticoides por vía oral. Según las guías de consenso japonesas, está indicado tratar a aquellos pacientes que tengan ictericia obstructiva, dolor abdominal o de espalda o manifestaciones extrapancreáticas sintomáticas39. Aunque hay casos de resolución espontánea, dada la mayor tasa de recurrencia, se mantiene la recomendación de tratar para evitar las consecuencias irreversibles de la fibrosis e inflamación7,40.
Antes de iniciar el tratamiento, es necesario asegurar el drenaje de la vía biliar en los pacientes con ictericia obstructiva y un buen control glucémico en caso de diabetes mellitus.
El fármaco de elección es la prednisolona oral, que inicialmente se debe administrar a dosis de 0,6mg/kg/día durante 2-3 semanas. Posteriormente debe hacerse un descenso gradual durante 2-3 meses (5mg cada 1-2 semanas) hasta alcanzar la dosis de 2,5-5mg al día. La respuesta al tratamiento se debe valorar de forma objetiva con la imagen radiológica del páncreas y/o de las manifestaciones extrapancreáticas. Si no se aprecia respuesta debe replantearse el diagnóstico de la PAI, buscando otros alternativas, especialmente el de neoplasia.
El tratamiento de mantenimiento con prednisolona (2,5-5mg/día) se intentará administrar durante un periodo máximo de 3 años. Algunos centros abogan por el tratamiento de mantenimiento libre de glucocorticoides, basado en inmunomoduladores como la azatioprina, a dosis de 2mg/kg/día durante un tiempo variable de 1 a 3 años. Otros optan por una estrategia sin esta fase, aunque la tasa de recurrencia en estos pacientes (53-60%) es significativamente mayor que la de aquellos que sí la hacen (40%)14,41-44. Factores que se han asociado con la recurrencia son la colangitis esclerosante con afectación del árbol biliar proximal, el aumento difuso de la glándula pancreática, la asociación con otras manifestaciones extrapancreáticas o la elevación sérica de IgG411,44-46.
En caso de recurrencia, se debe iniciar un nuevo ciclo de corticoides, planteando un descenso progresivamente más lento47. Si hay recaída durante el tratamiento de mantenimiento, se pueden administrar bolos adicionales de metilprednisolona (2 ciclos de 500mg/día, 3 días por semana) o un tratamiento concomitante con inmunomoduladores (azatioprina 1-2,5mg/kg/día)7,48,49. Otros fármacos empleados son el micofenolato mofetil, la ciclosporina, el metotrexato, la 6-mercaptopurina, la ciclofosfamida o el rituximab11.
PronósticoAunque hay casos de PAI con desarrollo de neoplasia pancreática, no hay estudios sólidos que permitan establecer esta asociación. Sin embargo, la baja incidencia de esta entidad, hace difícil la realización de estudios prospectivos con un tamaño muestral suficiente para definir esta relación.
ConclusiónLa PAI es una enfermedad de carácter benigno, de la que quedan todavía muchos aspectos por conocer. Existen 2 tipos de PAI con diferencias clínicas e histológicas bien definidas, cuyo principal rasgo en común es la respuesta al tratamiento con corticoides.
La relevancia clínica de esta entidad radica en ser parte del diagnóstico diferencial de la neoplasia pancreática ya que a pesar de poder manifestarse de una forma similar, tanto clínica como radiológicamente, el pronóstico es radicalmente distinto. Distinguir ambas entidades conlleva, además de una buena anamnesis, la realización de diferentes pruebas complementarias como pruebas de laboratorio, radiológicas, endoscópicas e histológicas. La obtención de material histológico, aunque no es imprescindible, es una parte clave en el algoritmo diagnóstico. Existe controversia en cuanto a la obtención y estudio del material hitológico/citológico, por lo que son necesarios más ensayos clínicos para aclarar estos aspectos. Ante una duda diagnóstica bien argumentada, con pruebas negativas para cáncer, la prueba terapéutica con corticoides durante 2 semanas puede esclarecer el diagnóstico de esta entidad. Sin embargo, esto no debe demorar el diagnóstico de una neoplasia pancreática más allá de estos 15 días.
En conclusión, la PAI es una entidad poco frecuente que es necesario conocer, ya que requiere para su diagnóstico de un alto índice de sospecha por parte del clínico. Sin embargo, dada la mayor incidencia de adenocarcinoma de páncreas y su pronóstico desfavorable, es necesario tener siempre la neoplasia pancreática como primera posibilidad cuando se plantee el diagnóstico diferencial ante un paciente con síntomas biliopancreáticos.
FinanciaciónNo ha habido fuentes de financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

