




En los últimos años, el estudio bioquímico y molecular de los diversos tipos de hemocromatosis ha llevado a la certeza de que el péptido hepcidina es el regulador central de la absorción de hierro. Este péptido que se sintetiza en el hígado ejerce su función a través de la degradación de la proteína ferroportina. La ferroportina es una bomba biológica de hierro situada en el epitelio intestinal y en la membrana de los macrófagos, su función es la de transportar hierro desde la célula intestinal al plasma y desde el macrófago al eritrón. En la hemocromatosis se produce un déficit físico o funcional de la hepcidina que conduce a un incremento de la ferroportina y, con esto, a una absorción de hierro excesiva. En situaciones de inflamación sucede lo contrario, se estimula la síntesis de hepcidina y se bloquea la entrada de hierro en el organismo y la síntesis de hemoglobina.
In the last few years, biochemical and molecular study of the various types of hemochromatosis have established that the hepcidin peptide is the central regulator of iron absorption. This peptide, which is synthesized in the liver, acts through ferroportin degradation. Ferroportin is an iron exporter situated in the intestinal epithelium and in the macrophage membrane whose function is to transport iron from the intestinal cell to plasma and from the macrophage to the erythron. In hemochromatosis, there is a physical or functional hepcidin deficit that increases ferroportin, thus producing excessive iron absorption. The opposite occurs in situations of inflammation: hepcidin synthesis is stimulated while iron entry into the organism and hemoglobin synthesis are blocked.
En la actualidad, se considera a la hepcidina como el factor de regulación central de la absorción intestinal de hierro así como del tránsito de este metal a través de la membrana del macrófago. Este péptido, compuesto por 25 aminoácidos, se aísla inicialmente en orina1. Presenta actividad antibacteriana y antifúngica2 de características similares a péptidos aislados en el órgano graso (órgano antecesor del hígado) en insectos (drosomicina en la drosófila). En los vertebrados, la síntesis de este péptido es hepática y proviene de la escisión de la proteína original, codificada por un gen situado en el cromosoma 19 y formado por 3 exones y 2 intrones. Esta escisión conlleva la formación de 3 péptidos de 25, 22 y 20 aminoácidos, de los que sólo se reconoce actividad en el de 25 aminoácidos. Químicamente, su característica más relevante es la presencia de 8 residuos del aminoácido cisteína, que forman 4 puentes disulfuro entre éstas, lo que le confiere una estructura tridimensional peculiar.
La primera pista que condujo al esclarecimiento de las funciones de la hepcidina en el metabolismo férrico la consiguió Pigeon et al3, al advertir que la síntesis hepática del ácido ribonucleico (ARN) codificante de este péptido se incrementaba de forma muy notable en un modelo de ratón con sobrecarga de hierro. Posteriormente, Nicolas et al4,5 advirtieron que en los ratones knockout para el gen UDF2 (relacionado con la diabetes) se producía una tremenda sobrecarga férrica. Los investigadores analizaron las secuencias genéticas dañadas por el procedimiento y comprobaron que en el proceso no sólo se había dañado al gen UDF2, sino también al gen codificante de la hepcidina, que se hallaba en una región genómica muy cercana. Además, estos investigadores comprobaron que estos ratones hemocromatósicos no eran capaces de sintetizar ARN correspondiente al gen de hepcidina. Estas investigaciones sentaron las bases para establecer una hipótesis según la que la hepcidina podía ser un potente regulador negativo de la absorción de hierro. Esta hipótesis explica el incremento de su síntesis en situaciones de sobrecarga férrica, que frena la absorción de hierro (precisamente para evitar una sobrecarga mayor). Al mismo tiempo predice que la ausencia de hepcidina (como, por ejemplo, sucede en el modelo de ratón knockout) determinará un incremento notable en la absorción intestinal del hierro. También se demostró en animales que la ferropenia, la anemia y la hipoxia, 3 situaciones que se benefician de la síntesis de hemoglobina (que a su vez necesita de una mayor absorción de hierro), son potentes inhibidores de la síntesis de hepcidina6.
Por otra parte, desde hace años es bien conocido que la inflamación se asocia a anemia y a secuestro férrico en el compartimento mononuclear y fagocítico, pero se desconocía el mediador de este fenómeno frecuentemente observado en la clínica diaria. Se demostró que los mediadores de la inflamación y, más concretamente, la interleucina 6, eran inductores potentes de la síntesis hepática de hepcidina. Resultó especialmente interesante la publicación de un caso clínico en que un paciente que presentaba un adenoma hepático que secretaba hepcidina en grandes cantidades experimentaba a su vez un cuadro clínico indistinguible de la anemia de proceso crónico (anemia inflamatoria), cuadro que revirtió al extirpar este adenoma. A partir de este dato se hipotetizó que la inflamación posiblemente conducía a la anemia de proceso crónico a través de la síntesis de hepcidina, una hipótesis que cada vez toma más fuerza. La hepcidina produciría un freno al paso de hierro desde el compartimento mononuclear y fagocítico al eritrón (bloqueo inflamatorio) con el consecuente déficit de hierro para la síntesis de hemoglobina y, en consecuencia, la presentación de anemia.
Un descubrimiento relevante en el esclarecimiento de las funciones de la hepcidina fue la demostración de que el receptor de la hepcidina era la recién descubierta proteína transportadora de hierro, ferroportina7. La ferroportina es una proteína con 12 dominios transmembrana que se halla fundamentalmente en la membrana laterobasal del epitelio intestinal y en la membrana macrofágica. Se ha demostrado que es la causante fisiológica del bombeo de hierro en la absorción intestinal y en la cesión del hierro almacenado en macrófagos al eritrón. Se demostró que la unión de la hepcidina con la ferroportina produce la internalización y la degradación enzimática de ésta última, frenando la acción de transporte de ésta. Los trabajos recientes han definido con precisión cómo se produce esta interacción y qué pasos bioquímicos conducen a la degradación celular de la ferroportina mediada por hepcidina8. En concreto, la hepcidina induce la fosforilación de los aminoácidos localizados en el lazo intracelular de la ferroportina, lo que desencadena con esto la internalización del complejo ferroportina–hepcidina, un hecho que conduce a la ubiquitinización de esta proteína y a la degradación lisosómica de ambas. Con este descubrimiento se comprendió por qué un incremento en la concentración de hepcidina, ya sea por sobrecarga férrica o inflamación, conlleva un bloqueo en la absorción de hierro (por destrucción de la ferroportina intestinal) y, a su vez, un bloqueo de la cesión del hierro del sistema mononuclear fagocítico (SMF) al eritrón (por destrucción de la ferroportina de la membrana del macrófago) con la aparición de anemia inflamatoria. La situación opuesta favorece la función de la ferroportina y, con esto, la absorción de hierro intestinal y la cesión de hierro del SMF al eritrón.
Dado que no hemos sido evolutivamente dotados de mecanismos de excreción del hierro, todo el control de la cantidad de hierro en el organismo recae en la regulación de la absorción intestinal del metal. Por esto, no resulta extraño que las anomalías en el control de la absorción de hierro puedan redundar en ferropenia o hemocromatosis. Dado que este control se traduce fundamentalmente en la acción de la hepcidina, es lógico suponer (y efectivamente así es) que los déficits de este regulador supongan la presencia de hemocromatosis y su exceso de anemia.
En la actualidad se distinguen 5 tipos genéticos diferentes de hemocromatosis hereditaria: 4 con herencia recesiva y uno dominante. Todos éstos comparten la misma fisiopatología, que consiste en una absorción intestinal de hierro anormalmente elevada en relación con los depósitos ya incrementados de hierro corporal. Los 4 tipos recesivos se deben a mutaciones de los genes que codifican las proteínas HFE9, hepcidina10, hemojuvelina (HJV)11 y receptor 2 de la transferrina12, y los 4 comparten un inapropiadamente bajo valor de hepcidina urinaria y plasmática para la cantidad de hierro corporal11,13–19. Además, la gravedad de la enfermedad parece correlacionarse con este desajuste, es decir, cuanto más bajo es el valor de hepcidina, mayor es la sobrecarga férrica y, por tanto, la gravedad de la enfermedad. Esta relación resultó evidente cuando se demostró que una pequeña proporción (5%) de los tipos más graves de hemocromatosis hereditaria de rápida aparición y progresión, la llamada hemocromatosis juvenil, era debida a la presencia de mutaciones recesivas del gen de la hepcidina que se asociaban a una pérdida de función del péptido10. Otra característica común a todas las formas de hemocromatosis hereditaria recesiva es que en todos los casos se aprecia sorprendentemente deficiencia de hierro en los macrófagos a pesar de la sobrecarga del hierro corporal. Por tanto, la sobrecarga parenquimatosa de hierro que coexiste con deficiencia férrica del compartimento mononuclear–fagocítico es un rasgo anatomopatológico que puede considerarse patognomónico del fenotipo por déficit de hepcidina y sobreexpresión de ferroportina típicamente presente en la hemocromatosis hereditaria de carácter recesivo. Sin embargo, el fenotipo de la hemocromatosis hereditaria autosómica dominante, ligada a mutaciones del gen de la ferroportina, es más variable. Los casos de comportamiento clínico similares a los descritos para las causas recesivas se caracterizan por presentar una ferroportina insensible a la regulación por hepcidina20–23 y, por tanto, se comportan como si el valor de hepcidina fuese bajo, aunque no lo sea. Por tanto, parece que la desregulación del eje hepcidina–ferroportina explica la mayor parte, si no todas, las características clínicas y anatomopatológicas de la enfermedad.
Ha sido más complicado desentrañar los mecanismos reguladores de la síntesis de hepcidina. En el esclarecimiento de estos mecanismos ha resultado fundamental el estudio de los casos de hemocromatosis juvenil ligados a mutaciones del gen de la HJV. Este tipo de alteración explica la mayor parte de los casos de esta enfermedad (95%), y en todos estos se ha demostrado un valor bajísimo de hepcidina en orina sin estar directamente afectado el gen de la hepcidina. Esto llevó a pensar que el producto del gen de la HJV resultaba crítico en la regulación de la síntesis de hepcidina, como posteriormente se ha demostrado.
La HJV es una proteína de la membrana celular, que se une al fosfatidil inositol glucosilado. Sus lugares de expresión son el músculo esquelético y cardíaco y los hepatocitos11. Se trata de una proteína homóloga a otras expresadas en el sistema nervioso central, que ejercen una acción en el crecimiento neuronal y actúan como correceptores para las denominadas BMP (bone morphogenetic proteins ‘proteínas morfogenéticas óseas’). La unión de las BMP con un correceptor desencadena una compleja cascada de acontecimientos bioquímicos que terminan con la translocación al núcleo celular y la estimulación de la transcripción genética mediante la unión a elementos del ácido desoxirribonucleico (ADN) que responden a las BMP. Cada vez parece más evidente que la proteína HJV es un correceptor de BMP que puede actuar como señal inductora de la transcripción del gen de la hepcidina en los hepatocitos o en las líneas celulares hepatocíticas24–27. De hecho, se ha demostrado que la infusión de BMP-2 en el ratón estimula la expresión de ARN mensajero de hepcidina e induce hipoferremia y, por tanto, corrobora la operatividad de esta cadena de acontecimientos metabólica in vivo26. Recientemente se ha descubierto que hay una forma de HJV soluble que procede de la proteólisis de la HJV unida a membranas y que parece presentar capacidad de interacción con BMP pero en sentido antagónico a la HJV unida a membranas28–30 o bien es posible que esta acción «antagónica» simplemente denote la pérdida de función de la HJV al dejar de estar unida a la membrana. Parece que las secuencias de ADN que median en la respuesta a BMP y HJV se localizan de 1,6 Kb a 2Kb previo a la zona de inicio de la transcripción del gen de la hepcidina31.
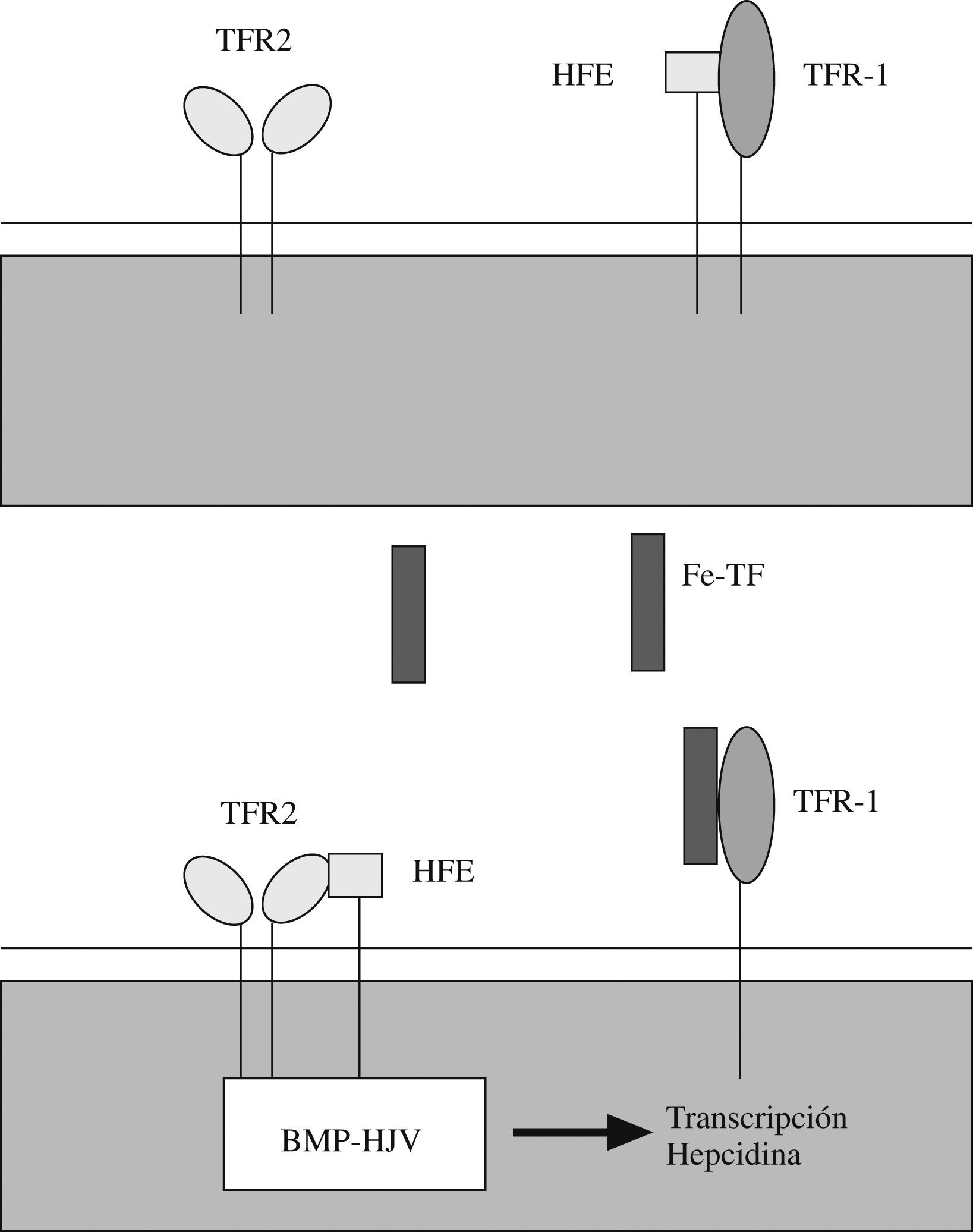
Se ha observado que la exposición in vitro de hepatocitos a un medio con elevada saturación de transferrina se traduce en un incremento de la señal BMP dependiente de HJV y, a su vez, de la síntesis de hepcidina25,32,33. Esto indica que la vía de señal BMP está regulada por la concentración de hierro. El mecanismo por el que la holotransferrina (transferrina diférrica) regula la señal BMP parece que comprende una serie de complejos multiproteicos y una serie de interacciones reguladoras competitivas que se dan en las superficies de membrana y que incluyen al receptor de la transferrina 1 (TFR 1), a la proteína ligada a la hemocromatosis HFE y al receptor de transferrina 2 (TFR 2).
La hemocromatosis hereditaria de tipo i o clásica comprende en España al 70% de los casos de hemocromatosis hereditaria. Se debe a mutaciones del gen codificante de la proteína HFE, una molécula de membrana similar a las proteínas HLA de tipo i34. Este tipo de hemocromatosis se asocia a déficits más leves de hepcidina y, por tanto, a una presentación clínica más leve que otras formas de hemocromatosis15,35. Parece ser que la proteína HFE tiene afinidad de unión con TFR 1 en la superficie celular, pero también con TFR 2 (aunque menos). La proteína HFE se mantiene fundamentalmente unida a TFR 1 mientras no haya una cantidad importante de transferrina diférrica unida a TFR 1, ya que esta unión desestabiliza la unión de TFR 1 con la HFE y desplaza a la HFE que tiende entonces a unirse a TFR 2. Es muy probable que la interacción entre HFE y TFR 2 desencadene la vía HJV–BMP, ya que se ha demostrado que TFR 2 puede asociarse a la proteína HJV. Por todo esto, parece que la ligazón entre el hierro y la vía de señal BMP dependiente de HJV se apoya en las interacciones competitivas presentes entre las proteínas HFE, TFR 1, TFR 2 y la holotransferrina y en una capacidad de regulación positiva de TFR 2 sobre la HJV36–44 (fig. 1). Esta cadena de acontecimientos explica que las mutaciones que afectan a HFE, TFR 2, HJV o hepcidina puedan potencialmente causar hemocromatosis.

Los conocimientos obtenidos en los últimos años respecto al mecanismo de control del hierro corporal ligado a la síntesis de hepcidina han transformado completamente el concepto que se tenía de la hemocromatosis hereditaria. En la actualidad se podría considerar la hemocromatosis hereditaria como una enfermedad genética de carácter endocrino (por déficit o disfunción de la hepcidina) similar a, por ejemplo, la diabetes. Como en la diabetes, el déficit de este elemento regulador (la insulina en el caso de la diabetes) conlleva valores elevados de un elemento (glucosa en un caso, hierro en otro) que producen daño parenquimatoso crónico. No resulta descabellado pensar que un día podrán tratarse muchos casos de hemocromatosis con análogos de la hepcidina. Sin embargo, la utilidad de la hepcidina puede concretarse antes en el terreno diagnóstico. En la actualidad, los métodos de laboratorio capaces de medir hepcidina en suero o en orina no son transportables a la práctica clínica, pero es de prever que lo serán en un período razonable y entonces probablemente se asistirá a una auténtica revolución en los métodos de diagnóstico de los trastornos por sobrecarga y quizá también por déficit de hierro.
Este trabajo ha sido posible en parte gracias al soporte de las becas del Fondo de Investigaciones Sanitarias (PI-04/1120) y de la Agencia d’Avaluació de Tecnología i Recerca Mèdica (005/29/2004).

