







The use of Janus kinase (JAK) inhibitors is a new approach in the therapy of inflammatory diseases with immune base. Tofacitinib is one of these inhibitors targeting JAK1 and JAK3, and its efficacy has been demonstrated in the treatment of moderate to severe ulcerative colitis (UC). It is a small synthetic molecule administered orally, with a fast bioavailability and elimination rate, predictable pharmacokinetics and lack of immunogenicity, which are convenient characteristics for both efficacy and safety. This article reviews the pharmacological characteristics of tofacitinib and its safety profile.
La inhibición de las quinasas Janus constituye un nuevo abordaje para el tratamiento de las enfermedades inflamatorias con base inmunitaria. Tofacitinib es un inhibidor preferente de las quinasas Janus 1 y 3, y su eficacia ha sido demostrada en el tratamiento de la colitis ulcerosa (CU) de moderada a grave. Se trata de una molécula pequeña sintética, de administración oral, con buena biodisponibilidad y eliminación rápida, farmacocinética predecible y ausencia de inmunogenicidad, características muy atractivas tanto para su eficacia como para su seguridad. En este artículo se revisan las cualidades farmacológicas de tofacitinib y su perfil desde el punto de vista de la seguridad.
Ulcerative colitis (UC) is a chronic, relapsing inflammatory bowel disease (IBD) characterised by alternating periods of activity and remission which can have a detrimental effect on quality of life.1 In Spain, the EpidemIBD study has shown the current incidence of UC to be 7.1 per 100,000 population per year.2
Current treatments for UC include aminosalicylates, corticosteroids, thiopurines, tacrolimus, ciclosporin and various biologic drugs. Aminosalicylates (5-ASA), the first-line treatment for UC, have an excellent safety profile. Corticosteroids are prescribed in the event of non-response or moderate-to-severe acute flare-ups, although their use should be limited to no more than 2–3 months, in order to limit side effects.3 The most widely used immunosuppressants in UC are thiopurines, which include 6-mercaptopurine and azathioprine. However, they have a high incidence of adverse effects/intolerance, requiring withdrawal of treatment in 20% of patients. Their immunomodulatory effect may also be related to the development of infections or cancer.3 Ciclosporin and tacrolimus are only used in severe, acute steroid-refractory colitis and require monitoring of drug concentrations in the blood. Their adverse effects include opportunistic infections, nephrotoxicity and neurotoxicity.4
Biologic drugs are proteins which act on extracellular cytokines or integrins; the most commonly used are tumour necrosis factor (TNF) inhibitors (infliximab, adalimumab or golimumab) and adhesion molecule inhibitors or anti-integrins (vedolizumab). However, TNF inhibitors can be associated with an increased risk of infections and cancer. Also, as they are protein-based and large in size, there is a risk of developing neutralising antibodies, which can lead to loss of efficacy or hypersensitivity reactions.5
Of the UC patients who do not respond to conventional therapy (aminosalicylates or corticosteroids), fewer than 50%–60% respond to thiopurines; the clinical remission rate with any of the currently available biologic agents is below 50%.6 As a lack or loss of response to a first biologic agent reduces the likelihood of response to a second or third,7 and the only option in these patients is proctocolectomy, it is clear that new treatment options are needed.
Recently, a large number of active substances called small molecule inhibitors that act on signal transduction, i.e. inside the cell, were developed. These drugs include tofacitinib, a Janus kinase (JAK) inhibitor.8 Tofacitinib was authorised in 2012 by the Food and Drug Administration9 for the treatment of rheumatoid arthritis (RA) and in 2017 by the European Medicines Agency (EMA), being extended to include active psoriatic arthritis and active UC in 2018.10
The purpose of this article is to review the pharmacological aspects and the safety profile of tofacitinib in UC patients, based on the data from the clinical development programme studies and their follow-up. It also briefly discusses the safety data obtained after the drug’s extended use in patients with RA, both in clinical trials and in registry studies.
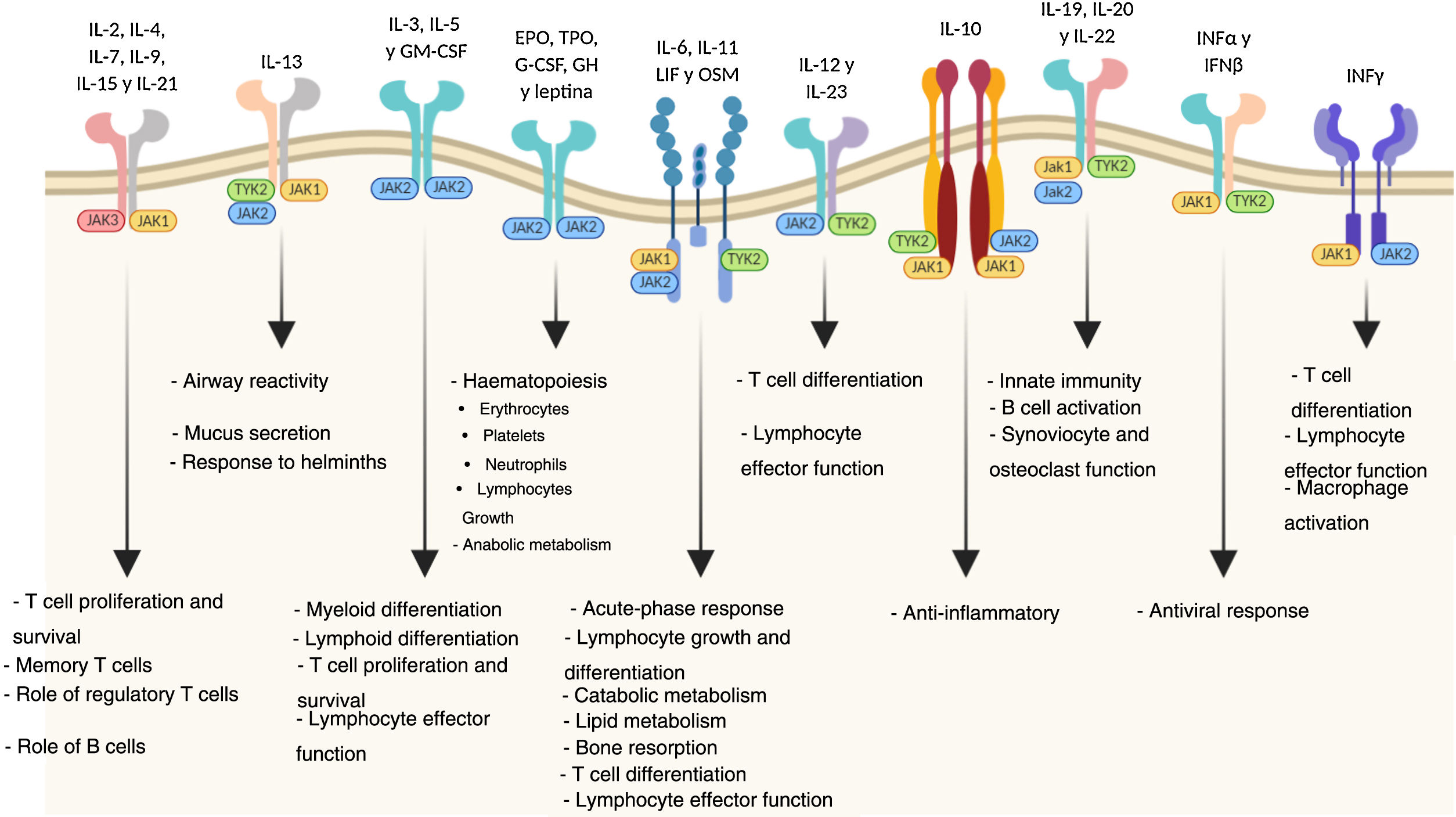
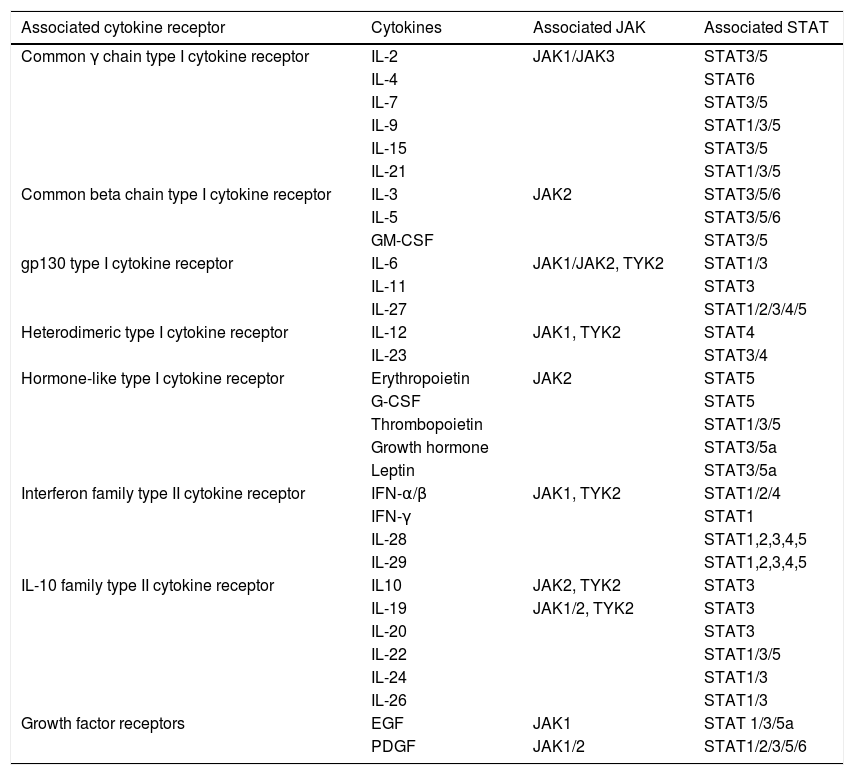
Mechanism of action of tofacitinibJanus kinases are at the root of various immune and inflammatory responses and include four intracellular tyrosine-kinase enzymes (JAK1, JAK2, JAK3 and TYK2 [tyrosine kinase-2]) associated with the intracellular membrane-bound region of different receptors which convert extracellular signals, mediated by different cytokines or hormones, into intracellular processes.11 The binding of a cytokine to the receptor causes its dimerisation, inducing activation of the associated JAK. When this happens, JAKs phosphorylate specific residues in the receptor’s cytoplasmic domain, where signal transducers and activators of transcription (STAT) reside. This is followed by phosphorylation of the STAT which, once activated, dimerise and translocate to the nucleus, where they play a role in the expression of multiple proteins.12,13 JAKs normally act in pairs made up of different JAKs, although JAK2 can also combine with itself. Each JAK pair is specific for a different group of cytokines (Table 1) and so activates different transduction signals which induce particular responses to various stimuli.14
JAK-STAT signalling pathways.
| Associated cytokine receptor | Cytokines | Associated JAK | Associated STAT |
|---|---|---|---|
| Common γ chain type I cytokine receptor | IL-2 | JAK1/JAK3 | STAT3/5 |
| IL-4 | STAT6 | ||
| IL-7 | STAT3/5 | ||
| IL-9 | STAT1/3/5 | ||
| IL-15 | STAT3/5 | ||
| IL-21 | STAT1/3/5 | ||
| Common beta chain type I cytokine receptor | IL-3 | JAK2 | STAT3/5/6 |
| IL-5 | STAT3/5/6 | ||
| GM-CSF | STAT3/5 | ||
| gp130 type I cytokine receptor | IL-6 | JAK1/JAK2, TYK2 | STAT1/3 |
| IL-11 | STAT3 | ||
| IL-27 | STAT1/2/3/4/5 | ||
| Heterodimeric type I cytokine receptor | IL-12 | JAK1, TYK2 | STAT4 |
| IL-23 | STAT3/4 | ||
| Hormone-like type I cytokine receptor | Erythropoietin | JAK2 | STAT5 |
| G-CSF | STAT5 | ||
| Thrombopoietin | STAT1/3/5 | ||
| Growth hormone | STAT3/5a | ||
| Leptin | STAT3/5a | ||
| Interferon family type II cytokine receptor | IFN-α/β | JAK1, TYK2 | STAT1/2/4 |
| IFN-γ | STAT1 | ||
| IL-28 | STAT1,2,3,4,5 | ||
| IL-29 | STAT1,2,3,4,5 | ||
| IL-10 family type II cytokine receptor | IL10 | JAK2, TYK2 | STAT3 |
| IL-19 | JAK1/2, TYK2 | STAT3 | |
| IL-20 | STAT3 | ||
| IL-22 | STAT1/3/5 | ||
| IL-24 | STAT1/3 | ||
| IL-26 | STAT1/3 | ||
| Growth factor receptors | EGF | JAK1 | STAT 1/3/5a |
| PDGF | JAK1/2 | STAT1/2/3/5/6 |
EGF: epidermal growth factor; G-CSF: granulocyte colony-stimulating factor; GM-CSF: granulocyte-macrophage colony-stimulating factor; IFN: interferon; IL: interleukin; JAK: Janus kinase; PDGF: platelet-derived growth factor; STAT: signal transducer and activator of transcription; TYK: tyrosine kinase.
The pathogenesis of UC is multifactorial, but it is probably the result of an abnormal immune response to components of the microbiota in genetically susceptible people exposed to environmental triggers. The balance between pro- and anti-inflammatory mediators is altered in these cases. Therefore, blockage of the JAK/STAT pathway, as it would interfere with the actions of multiple cytokines at the same time, might more broadly regulate both the innate and adaptive immune responses and be effective in slowing down the chronic inflammatory process in UC.15,16
Tofacitinib is a potent inhibitor of the JAK family which interferes with JAK-STAT signalling by competing with ATP for binding to the JAK kinase domain. Studies in vitro have demonstrated a preferential inhibition of JAK1 and JAK3, with a lesser effect on JAK2.17 As a result, tofacitinib inhibits phosphorylation and activation of JAKs, preventing phosphorylation and activation of STAT and subsequent gene transcription. This causes lower production of cytokines and modulation of the immune response, both innate and adaptive.18
As a result of the role of the JAK pathways in cell growth, maturation and differentiation, as well as in haematopoiesis, it must be taken into account that their inhibition is also accompanied by a safety profile which may determine their clinical effectiveness in UC (Fig. 1).
Differences between tofacitinib and biologic therapiesDifferences in mechanism of action
Whereas biologic drugs cause major, long-lasting inhibition of the corresponding cytokine target, JAK inhibitors regulate the inflammatory response in a more gradual, reversible way.15 The partial and reversible inhibition of multiple cytokines with tofacitinib results in a different therapeutic profile from that of biologic drugs.
Differences in their pharmacokinetic propertiesThe fact that biologic agents are large proteins and tofacitinib is a small synthetic molecule means that there are substantial differences in their pharmacokinetics, with significant implications for their administration schedule and dosage.
Absorption and distribution. One advantage of tofacitinib is that it is administered orally; it can withstand gastric breakdown and can rapidly enter the systemic circulation.19 Tofacitinib is characterised by: good absorption, with or without food (with a bioavailability of 74%); rapid onset of its effect (peak plasma concentrations are reached in 30−60 min); and an increase in systemic exposure proportionate to the dose (Table 2). Steady-state plasma concentration is achieved in 24−48 h, with negligible accumulation after twice-daily dosing (2 BID).10 Age, gender, body weight (no dose adjustment required for weight) or initial disease severity do not appear to have any effect on the mean plasma concentration of tofacitinib.20 Tofacitinib moderately binds to plasma proteins, especially albumin, and in healthy individuals it does not affect glomerular filtration, renal plasma flow or creatinine clearance.10,21
Pharmacological characteristics of tofacitinib and biologic agents.
| Biologic agents | Small molecules | |
|---|---|---|
| Molecular weight | >1 kDa | <700 Da |
| Route of administration | Parenteral | Oral |
| Half-life | 9–25 days | 3 h |
| Bioavailability | 50%–100% (depending on route of administration) | 74% |
| Elimination | Protein catabolism | Hepatic metabolism and renal excretion |
Biologic drugs are administered parenterally (subcutaneously or intravenously). Plasma clearance can be influenced by several factors, such as weight, serum albumin, the inflammation caused by the disease itself, and the development of antibodies against the drug.22 The distribution of monoclonal antibodies in tissues is limited due to their large size and hydrophilic nature; for example, IV treatment with infliximab immediately causes a high systemic concentration of the drug. However, with antibodies administered subcutaneously, absorption is through the lymphatic system and maximum concentration is achieved several days later.
Biotransformation and elimination. Elimination of tofacitinib is rapid (the half-life is approximately 3 h) and studies in healthy volunteers show that 95% of the drug (up to 100 mg, single dose) is eliminated within 24 h.10 Where treatment needs to be discontinued due to adverse effects or situations such as pregnancy or surgery, this is an advantage, as clearance is so rapid. Tofacitinib elimination is 70% by hepatic metabolism and 30% by renal excretion. Its metabolism is mainly mediated by CYP3A4, with CYP2C19 playing a minor role.10 It metabolises into at least eight metabolites, but the pharmacological activity is due to the molecule of origin.23 Biologic drugs are eliminated more slowly by protein catabolism (the half-life of infliximab is 9–12 days, that of adalimumab and golimumab is 2 weeks, and that of vedolizumab is 25 days).22,24
Pharmacological interactions. Circulating levels of tofacitinib and exposure increase when administered concomitantly with potent CYP3A4 inhibitors (e.g. ketoconazole, itraconazole, voriconazole or clarithromycin) or when concomitant administration of one or more medicinal products results in moderate inhibition of CYP3A4 and potent inhibition of CYP2C19 (e.g. fluconazole). In these cases, it is recommended that the dose of tofacitinib be halved. In contrast, circulating levels of tofacitinib decrease when it is administered together with potent CYP inducers (e.g. rifampicin), with the subsequent reduction in or even loss of response, for which concomitant administration is not recommended when starting tofacitinib therapy.10 Due to the risk of additional immunosuppression, azathioprine, tacrolimus and ciclosporin are not recommended concomitantly with tofacitinib.9
Special situations. The safety and efficacy of tofacitinib in children under 18 have not been established. In older patients (65 and over), due to an increased risk of infections, tofacitinib should only be considered if no suitable alternative treatment is available.10 Unlike monoclonal antibodies, the dose of tofacitinib should be reduced in patients with severe kidney or moderate liver failure, and it is contraindicated in cases of severe liver failure.10
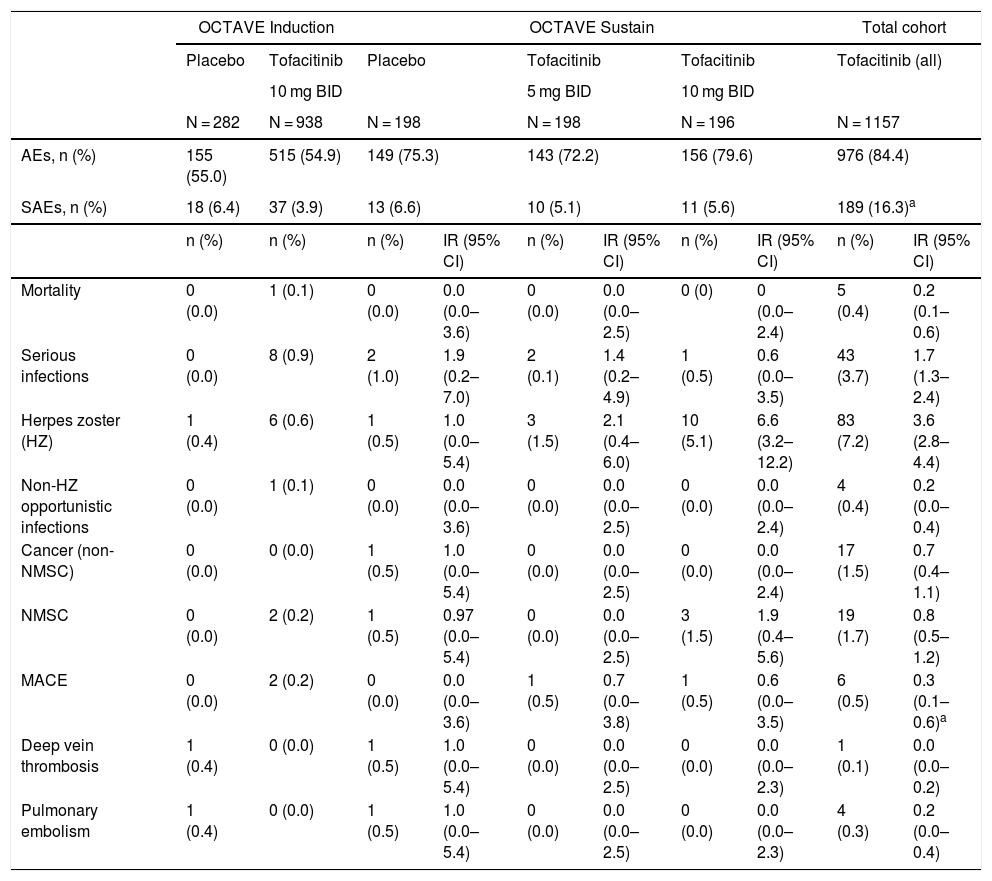
Safety of tofacitinib in the treatment of ulcerative colitisThe safety of tofacitinib in UC patients has been studied in a phase 2 clinical trial and in the phase 3 OCTAVE Induction 1 and OCTAVE Induction 2 studies, the OCTAVE Sustain maintenance study and the OCTAVE-Open extension study. The cohorts analysed are induction (8 weeks), maintenance (52 weeks) and total (including any patient who received tofacitinib 5 mg or 10 mg BID in any of the studies). The total duration of follow-up with tofacitinib has reached 6.1 years, and 1157 patients were included with 2403 patient-years of exposure. During the extension study, 83% of the patients received the dose of 10 mg BID; this is an important consideration, as some of the adverse effects of tofacitinib are dose-dependent. A general acceptable safety profile has been confirmed with both doses,20,25–27 and several meta-analyses support a safety profile of tofacitinib comparable to that of biologic drugs.28–30
In the induction groups (OCTAVE Induction 1 and 2), the proportion of patients with adverse events (AEs), serious adverse events (SAEs) and discontinuations due to AEs were similar in all treatment groups (Table 3). In the maintenance group, the proportion of patients with AEs and SAEs were also comparable.25 The most common AEs in patients taking tofacitinib 10 mg BID in induction studies were headache, nasopharyngitis, nausea and joint pain. In the induction and maintenance studies, in general, the most common categories of SAE were gastrointestinal disorders and infections, and the most common SAE was worsening of the UC.10
Safety outcomes during the clinical development of tofacitinib in UC. Data from the overall cohort include 6.1 years of follow-up.
| OCTAVE Induction | OCTAVE Sustain | Total cohort | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Placebo | Tofacitinib | Placebo | Tofacitinib | Tofacitinib | Tofacitinib (all) | |||||
| 10 mg BID | 5 mg BID | 10 mg BID | ||||||||
| N = 282 | N = 938 | N = 198 | N = 198 | N = 196 | N = 1157 | |||||
| AEs, n (%) | 155 (55.0) | 515 (54.9) | 149 (75.3) | 143 (72.2) | 156 (79.6) | 976 (84.4) | ||||
| SAEs, n (%) | 18 (6.4) | 37 (3.9) | 13 (6.6) | 10 (5.1) | 11 (5.6) | 189 (16.3)a | ||||
| n (%) | n (%) | n (%) | IR (95% CI) | n (%) | IR (95% CI) | n (%) | IR (95% CI) | n (%) | IR (95% CI) | |
| Mortality | 0 (0.0) | 1 (0.1) | 0 (0.0) | 0.0 (0.0–3.6) | 0 (0.0) | 0.0 (0.0–2.5) | 0 (0) | 0 (0.0–2.4) | 5 (0.4) | 0.2 (0.1–0.6) |
| Serious infections | 0 (0.0) | 8 (0.9) | 2 (1.0) | 1.9 (0.2–7.0) | 2 (0.1) | 1.4 (0.2–4.9) | 1 (0.5) | 0.6 (0.0–3.5) | 43 (3.7) | 1.7 (1.3–2.4) |
| Herpes zoster (HZ) | 1 (0.4) | 6 (0.6) | 1 (0.5) | 1.0 (0.0–5.4) | 3 (1.5) | 2.1 (0.4–6.0) | 10 (5.1) | 6.6 (3.2–12.2) | 83 (7.2) | 3.6 (2.8–4.4) |
| Non-HZ opportunistic infections | 0 (0.0) | 1 (0.1) | 0 (0.0) | 0.0 (0.0–3.6) | 0 (0.0) | 0.0 (0.0–2.5) | 0 (0.0) | 0.0 (0.0–2.4) | 4 (0.4) | 0.2 (0.0–0.4) |
| Cancer (non-NMSC) | 0 (0.0) | 0 (0.0) | 1 (0.5) | 1.0 (0.0–5.4) | 0 (0.0) | 0.0 (0.0–2.5) | 0 (0.0) | 0.0 (0.0–2.4) | 17 (1.5) | 0.7 (0.4–1.1) |
| NMSC | 0 (0.0) | 2 (0.2) | 1 (0.5) | 0.97 (0.0–5.4) | 0 (0.0) | 0.0 (0.0–2.5) | 3 (1.5) | 1.9 (0.4–5.6) | 19 (1.7) | 0.8 (0.5–1.2) |
| MACE | 0 (0.0) | 2 (0.2) | 0 (0.0) | 0.0 (0.0–3.6) | 1 (0.5) | 0.7 (0.0–3.8) | 1 (0.5) | 0.6 (0.0–3.5) | 6 (0.5) | 0.3 (0.1–0.6)a |
| Deep vein thrombosis | 1 (0.4) | 0 (0.0) | 1 (0.5) | 1.0 (0.0–5.4) | 0 (0.0) | 0.0 (0.0–2.5) | 0 (0.0) | 0.0 (0.0–2.3) | 1 (0.1) | 0.0 (0.0–0.2) |
| Pulmonary embolism | 1 (0.4) | 0 (0.0) | 1 (0.5) | 1.0 (0.0–5.4) | 0 (0.0) | 0.0 (0.0–2.5) | 0 (0.0) | 0.0 (0.0–2.3) | 4 (0.3) | 0.2 (0.0–0.4) |
AE: adverse event; CI: confidence interval; IR: incidence rate; MACE: major adverse cardiovascular event; NMSC: non-melanoma skin cancer; SAE: severe AE; UC: ulcerative colitis.
Out of the entire treated population there were five deaths. One of these people, who received tofacitinib 10 mg BID, died from a ruptured aortic aneurysm during the induction period, while the other four patients, who received tofacitinib 10 mg BID in the open-label extension study, died, respectively, of hepatic angiosarcoma, acute myeloid leukaemia, pulmonary embolism in the context of cholangiocarcinoma with peritoneal metastasis, and melanoma. The incidence rate (IR, expressed as patients with ≥1 event per 100 patient-years of exposure) of death was 0.2 (95% confidence interval [CI] 0.1−0.6).26
Infections, herpes zoster and tuberculosisInfections. In the OCTAVE Inductions 1 and 2 trials, serious infections were more common with tofacitinib (0.9%) than with placebo (0%). In OCTAVE Sustain study, the IR of severe infection was similar in the three treatment groups.20 In the total patient cohort up to 6.1 years treated with tofacitinib, the IR for severe infection was 1.75 (95% CI 1.27–2.36)27 (Table 3). None of the cases of severe infection in the clinical development programme in UC had a fatal outcome. Old age has been associated with an increased risk of opportunistic infections and herpes zoster (HZ), but not with serious infections.31 The EMA safety committee, after reassessing the data from the ongoing clinical study (A3921133) in patients aged 50 or over with rheumatoid arthritis and at least one cardiovascular risk factor, and the results of previous studies in all indications, found an increased risk of serious and fatal infections in patients over the age of 65.32
Herpes zoster. In the maintenance cohort, a higher IR (≥1 event per 100 patient-years of exposure) of HZ infection was found in patients treated with tofacitinib 5 mg BID (2.1) or 10 mg BID (6.6) than in the placebo group (1.0), the difference being statistically significant for the dose of 10 mg BID (Table 3). In other words, there is a dose-dependent association between treatment with tofacitinib and the risk of HZ. In the total cohort up to 6.1 years, the IR was 3.57 cases per 100 patient-years.27 In the safety analysis up to 4.4 years, many of the cases were classified as mild or moderate (96%), and there were only six cases of disseminated HZ and three cases of postherpetic neuralgia.33 In fact, 85% of the cases cleared up with antiviral treatment (up to the time of analysis), in most cases without the need to withdraw tofacitinib (44 out of 65 cases); in 16 patients, it was decided to temporarily discontinue the tofacitinib therapy. The independent risk factors for HZ infection were age (p < 0.0001) and previous failure with TNF inhibitors (p < 0.05), but not the length of time on treatment with tofacitinib.33
Opportunistic infections. Other opportunistic infections were uncommon in patients with UC treated with tofacitinib (Table 3). The IR (excluding HZ) in the total cohort of 1157 patients was 0.16 (95% CI 0.04–0.42), with no evidence of an increased risk of opportunistic infection with a longer duration of the treatment.27 The four cases which did occur consisted of cytomegalovirus colitis, pulmonary cryptococcosis, histoplasmosis and cytomegalovirus hepatitis.26,27
Although no cases of tuberculosis have been reported in induction and maintenance clinical trials with tofacitinib in UC,20 it is recommended that local guidelines regarding the detection of latent tuberculosis be followed.10
VaccinationPatients with IBD have a higher risk of severe infections and HZ, and this risk is increased by treatment with tofacitinib, biologic agents, thiopurines, corticosteroids and combined therapies.34–36 Therefore, it seems advisable to assess the need to vaccinate these patients as a preventive measure.37 It is also advisable to ensure that the patient is up to date on the vaccination schedule before starting treatment with tofacitinib, as indicated in the clinical guidelines.38 Concomitant administration of live vaccines with tofacitinib is not recommended. Immunisation with live vaccines should be done at least two weeks (although four is preferable) before the start of treatment with tofacitinib.10
The European Medicines Agency (EMA) has authorised an inactivated vaccine for recombinant HZ virus (Shingrix®), opening up the possibility of avoiding other vaccines with attenuated virus in susceptible patients. Although there are currently no definitive recommendations on vaccination against HZ in Europe, the Grupo Español de Trabajo en Enfermedad de Crohn y Colitis Ulcerosa [Spanish Working Group on Crohn’s Disease and Ulcerative Colitis], in its document on vaccines and algorithms for action in patients with IBD, recommends the use of Shingrix® for patients receiving immunosuppressive treatment.39 The Advisory Committee on Immunization Practices recommends the recombinant vaccine in all adults over the age of 50 and in patients with a history of HZ, patients with a history of chronic conditions and patients on immunosuppression. The latter two groups would include the vast majority of patients with IBD.40
NeoplasmsIn the total cohort of the integrated safety analysis (up to 6.1 years of follow-up), the IR for neoplasms (excluding non-melanoma skin cancer [NMSC]) was 0.69 (95% CI 0.4–1.11) (Table 3). Given that a proportion of these patients had also received other immunosuppressants, such as TNF inhibitors or thiopurines, the cause of these tumours may be debatable. There were 17 cases of cancer (two cases of cervical cancer, two of breast cancer, two of colon adenocarcinoma, one hepatic angiosarcoma, one cholangiocarcinoma, one cutaneous leiomyosarcoma, one diffuse large B-cell lymphoma, one Epstein–Barr virus-associated lymphoma, one melanoma, one renal cell carcinoma, one lung cancer, one invasive ductal carcinoma in situ of the breast, one essential thrombocythaemia and one acute myeloid leukaemia), plus secondary neoplasms in the liver and peritoneum.41
The IR for NMSC was 0.78 (95% CI 0.47–1.22) (Table 3); in the analysis up to 6.1 years, of the 19 patients with NMSC, 17 had been exposed to thiopurines and 15 had been exposed to TNF inhibitors. These IRs are similar to those of patients with UC treated with biologic drugs.41,42
Lipids and cardiovascular complicationsPatients treated with tofacitinib were found to have an increase in blood lipid concentration (total cholesterol [TC], low-density lipoprotein cholesterol [LDL-C] and low-density lipoprotein cholesterol [HDL-C]).20 Changes in lipid profile stabilised after 4–8 weeks of treatment but reverted after stopping treatment. TC/HDL-C and LDL-C/HDL-C ratios, which are better predictors of cardiovascular risk than individual lipids, remained unchanged.43 Overall, in the 4.4 years of follow-up with tofacitinib, 4.8% of the study population were prescribed a new lipid-lowering treatment and 1.5% had their dose of their lipid-lowering drugs increased.43 Lipid concentrations should be checked 4–8 weeks after the start of treatment10 (Table 4).
Recommendations for the clinical management of patients treated with tofacitinib.
| On starting | 4–8 weeks after starting | Every 3 months from then on | |
|---|---|---|---|
| Lymphocytes | |||
| Neutrophils | |||
| Haemoglobin | |||
| Lipids | |||
| Liver enzymes | It is also recommended that liver values be regularly monitored and that the reason for any elevation in liver enzymes be investigated immediately | ||
| Do not start treatment in patients with: | Lymphocyte count <750 cells/mm3 | Neutrophil count <1000 cells/mm3 | Haemoglobin levels <9 g/dl |

Created from the summary of product characteristics for Xeljanz®.
Serious cardiovascular complications were uncommon in the tofacitinib trials in patients with UC (IR 0.3; 95% CI 0.1–0.6).26 In the total cohort up to 4.4 years, four patients who received tofacitinib had a confirmed cardiovascular AE (major adverse cardiovascular event [MACE]): haemorrhagic stroke, aortic dissection, acute coronary syndrome and acute myocardial infarction.25 Three of the four patients had four or more baseline risk factors, including hyperlipidaemia.43
Thromboembolic eventsPatients with inflammatory diseases have an increased risk of deep vein thrombosis (DVT) and pulmonary embolism (PE).44 In UC, the IRs are 0.07–0.30 for DVT and 0.04–0.20 for PE.45 In the total cohort of the clinical development programme for tofacitinib in UC (≤6.1 years of follow-up), one patient had a DVT (IR 0.04; 95% CI 0.00−0.23) and four had a PE (IR 0.16; 95% CI 0.04–0.41). The predominant tofacitinib dose was 10 mg BID. All had risk factors for thromboembolism.45
Tofacitinib may increase the risk of venous thromboembolism (VTE), both DVT and PE, in high-risk patients (prior VTE, major surgery, immobilisation, myocardial infarction in the previous 3 months, heart failure, use of combined hormonal contraceptives or hormone replacement therapy, congenital bleeding disorder or malignant neoplasm); other additional risk factors for VTE include advanced age, obesity, diabetes, hypertension and smoking.32 Regardless of the indication and dose, tofacitinib should be used with caution in all patients with risk factors for VTE.
Assessment of the data suggests that the risk of VTE is higher in patients taking tofacitinib 10 mg BID and in those treated over a long period. Therefore, maintenance doses of 10 mg BID are not recommended in patients with UC at risk of VTE, unless there is no suitable alternative treatment.10,32,46 All patients treated with tofacitinib should be monitored for signs and symptoms suggestive of VTE and told to seek immediate medical attention if they experience such symptoms.32,46
Laboratory testsDuring the induction studies, levels of neutrophils, lymphocytes and haemoglobin remained stable. In the maintenance study, the IRs for anaemia were 2.91, 5.51 and 2.55, respectively, for the placebo, tofacitinib 5 mg BID and tofacitinib 10 mg BID groups. The IRs for neutropenia were 0, 0.67 and 0.64, respectively, while no lymphopenia was reported.47 With the exception of lipids, no significant changes in laboratory values were noted from the beginning of the open-label extension phase to 36 months of follow-up.48 Nevertheless, complete blood count monitoring in accordance with the recommendations shown in Table 4 is essential.
PregnancyThe use of tofacitinib during pregnancy and lactation is contraindicated.10 There have been some cases, however, of exposure to tofacitinib in mothers (n = 74) or fathers (n = 84) at the time of conception or before or during pregnancy in studies with tofacitinib for all indications.49 In those with UC, 11 cases of maternal exposure and 14 cases of paternal exposure to tofacitinib have been identified. The outcomes included 15 healthy newborns, no fetal deaths, no neonatal deaths, no congenital malformations, two miscarriages and two induced abortions. Although the data are very limited, these outcomes appear similar to those published on other populations exposed to tofacitinib and those of the general population.49
Safety of tofacitinib in patients with rheumatoid arthritisTofacitinib was first tested and authorised for the treatment of RA, so there are more long-term data on its safety in RA than in the indication for UC. In the integrated safety analysis of tofacitinib in 7061 patients with RA (22,875 patient-years of exposure) up to 9.5 years with a median exposure of 3.1 years and 30% of patients having more than 5 years of exposure, the IR (expressed as patients with ≥1 event per 100 patient-years of exposure) of SAEs was 9.0 (95% CI 8.6–9.4).50 There were serious infections in 576 patients (8.2%; IR 2.5; 95% CI 2.3–2.7), and 782 patients (11.1%) developed HZ (IR 3.6; 95% CI 3.4–3.9). Neoplasms (excluding NMSC) occurred in 177 patients (2.5%; IR 0.8; 95% CI 0.7−0.9), NMSC in 129 patients (1.8%; IR 0.6; CI 95% 0.5–0.7) and lymphoma in 12 patients (0.2%; IR 0.1; 95% CI 0.0–0.1). A total of 85 patients (1.3%) had a MACE (IR 0.4; 95% CI 0.3−0.5), 27 (0.4%) had VTE (IR 0.12; 95% CI 0.08–0.17) and 28 (0.4%) had a PE (IR 0.12; 95% CI 0.08–0.17). Fifty-nine (59) patients died (0.8%; IR 0.3; 95% CI 0.2–0.3).50
The ORAL Surveillance study (A3921133) is an ongoing, post-marketing safety study with a large sample size (n = 4362) comparing tofacitinib with TNF inhibitors in patients with RA aged 50 and over and having at least one cardiovascular risk factor. Patients were randomised to receive tofacitinib 10 mg BID, 5 mg BID or a TNF inhibitor (etanercept 50 mg once a week or adalimumab 40 mg every 2 weeks). The IR of PE was statistically higher in patients treated with tofacitinib 10 mg BID (IR 0.54; 95% CI 0.32−0.87) compared with that in patients treated with TNF inhibitors (IR 0.09; 95% CI % 0.02−0.26). The IR for the dose of 5 mg BID (0.27; 95% CI 0.12−0.52) was higher than that of the TNF inhibitors, but the difference was not statistically significant.32,51 The risk of serious infections (fatal and non-fatal) increased more in patients over 65 than in younger patients.10 With regard to mortality, there were no statistically significant differences with 5 mg BID, but there were between 10 mg BID (IR 0.89; 95% CI 0.59–1.29) and TNF inhibitors (IR 0.27; 95% CI 0.12−0.51).32,51
ConclusionThe safety of tofacitinib has been studied in patients with moderate to severe UC, in induction and maintenance therapy, with a follow-up in clinical trials of up to 6.1 years. The safety profile observed in these trials is acceptable and manageable, although additional data from clinical practice are needed to confirm this.
When prescribing tofacitinib, it is necessary to take into account, on the one hand, the significant adverse effects that occur during most treatments for UC and, on the other hand, the benefits of tofacitinib, such as its oral administration, lack of immunogenicity and short half-life.
Conflicts of interestAntonio López-Sanromán has been a lecturer for Pfizer and has participated in discussion sessions on tofacitinib and ulcerative colitis. Juan V. Esplugues has received fees for giving lectures and expert advice and grants to conduct research projects from the following companies: MSD, AbbVie, AstraZeneca, Gilead and Pfizer. Eugeni Domènech has received fees for giving lectures and expert advice and has had entrance fees paid for courses and conferences, and his unit has received donations or grants for holding events or conducting research projects from the following companies: MSD, AbbVie, Takeda, Kern Pharma, Pfizer, Janssen, Celgene, Otsuka Pharmaceuticals, Adacyte, Ferring, Shire Pharmaceuticals, Tillots, Thermofisher, Grifols and Gebro.
The authors would like to thank Mónica Valderrama, Ana Cábez and Susana Gómez from Pfizer for the literature review and revision of this manuscript. They would also like to thank Dr Ana Moreno Cerro and Anabel Herrero PhD for their help, on behalf of Springer Healthcare and funded by Pfizer España, with the drafting of the manuscript.
Please cite this article as: López-Sanromán A, Esplugues JV, Domènech E. Farmacología y seguridad de tofacitinib en colitis ulcerosa. Gastroenterol Hepatol. 2021;44:42–49.
