







La inhibición de las quinasas Janus constituye un nuevo abordaje para el tratamiento de las enfermedades inflamatorias con base inmunitaria. Tofacitinib es un inhibidor preferente de las quinasas Janus 1 y 3, y su eficacia ha sido demostrada en el tratamiento de la colitis ulcerosa (CU) de moderada a grave. Se trata de una molécula pequeña sintética, de administración oral, con buena biodisponibilidad y eliminación rápida, farmacocinética predecible y ausencia de inmunogenicidad, características muy atractivas tanto para su eficacia como para su seguridad. En este artículo se revisan las cualidades farmacológicas de tofacitinib y su perfil desde el punto de vista de la seguridad.
The use of Janus kinase (JAK) inhibitors is a new approach in the therapy of inflammatory diseases with immune base. Tofacitinib is one of these inhibitors targeting JAK1 and JAK3, and its efficacy has been demonstrated in the treatment of moderate to severe ulcerative colitis (UC). It is a small synthetic molecule administered orally, with a fast bioavailability and elimination rate, predictable pharmacokinetics and lack of immunogenicity, which are convenient characteristics for both efficacy and safety. This article reviews the pharmacological characteristics of tofacitinib and its safety profile.
La colitis ulcerosa (CU) es una enfermedad inflamatoria intestinal (EII) crónica y recidivante, que cursa con periodos de actividad y de remisión, y que puede llegar a afectar negativamente a la calidad de vida1. En España, los resultados del estudio EpidemIBD muestran una incidencia actual de la CU de 7,1 por 100.000 habitantes-año2.
Las terapias actuales para la CU incluyen aminosalicilatos, corticoides, tiopurinas, tacrolimus, ciclosporina y diversos fármacos biológicos. Los aminosalicilatos (5-ASA), primera línea de tratamiento de la CU, tienen un perfil de seguridad excelente. En caso de no respuesta o de brote agudo moderado o grave se prescriben corticoides, aunque su uso debe limitarse a períodos no superiores a 2-3 meses, para limitar los efectos adversos3. Los inmunosupresores más utilizados en la CU son las tiopurinas, que incluyen la 6-mercaptopurina y la azatioprina, pero presentan una considerable incidencia de efectos adversos/intolerancia que obliga a la retirada del tratamiento en el 20% de los pacientes. Su efecto inmunomodulador puede relacionarse además con la aparición de infecciones o tumores3. Ciclosporina y tacrolimus se utilizan solo en caso de colitis aguda grave corticorrefractaria y requieren monitorización de la concentración del fármaco en sangre. Entre sus efectos adversos destacan las infecciones oportunistas, la nefrotoxicidad y la neurotoxicidad4.
Los medicamentos biológicos son proteínas que actúan sobre citocinas o integrinas extracelulares, siendo los más empleados los inhibidores del factor de necrosis tumoral (infliximab, adalimumab, golimumab; iTNF) y los inhibidores de las moléculas de adhesión o antiintegrinas (vedolizumab). Sin embargo, los iTNF pueden asociarse con un mayor riesgo de infecciones y tumores. Además, por su carácter proteico y su gran tamaño, existe el riesgo de desarrollo de anticuerpos neutralizantes que contribuirían a la pérdida de eficacia o a la aparición de reacciones de hipersensibilidad5.
De los pacientes con CU que no responden a la terapia convencional (aminosalicilatos, corticoides), menos del 50-60% responden a tiopurinas, siendo la tasa de remisión clínica con cualquiera de los agentes biológicos disponibles en la actualidad inferior al 50%6. Teniendo en cuenta que la falta o la pérdida de respuesta a un primer agente biológico reduce la probabilidad de respuesta a un segundo o tercero7, y que la única opción en estos pacientes es la proctocolectomía, es evidente que son necesarias nuevas alternativas terapéuticas.
Recientemente, se están desarrollando un gran número de principios activos denominados moléculas pequeñas inhibidoras (small molecules inhibitors), que actúan sobre la transducción de señales, es decir, en el interior de la célula. Entre estos fármacos se encuentra tofacitinib, un inhibidor de las quinasas Janus o JAK (de Janus kinases, en inglés)8. En 2012, tofacitinib fue autorizado por la Food and Drug Administration9 para el tratamiento de la artritis reumatoide (AR), y en 2017 por la European Medicines Agency (EMA), ampliándose para artritis psoriásica activa y CU activa en 201810.
El propósito de este artículo es revisar los aspectos farmacológicos y el perfil de seguridad de tofacitinib en pacientes con CU, a partir de los datos obtenidos en los estudios del programa de desarrollo clínico y su seguimiento. Además, se describirán brevemente los datos de seguridad obtenidos después de su dilatado uso en pacientes con AR, tanto en ensayos clínicos como en estudios de registro.
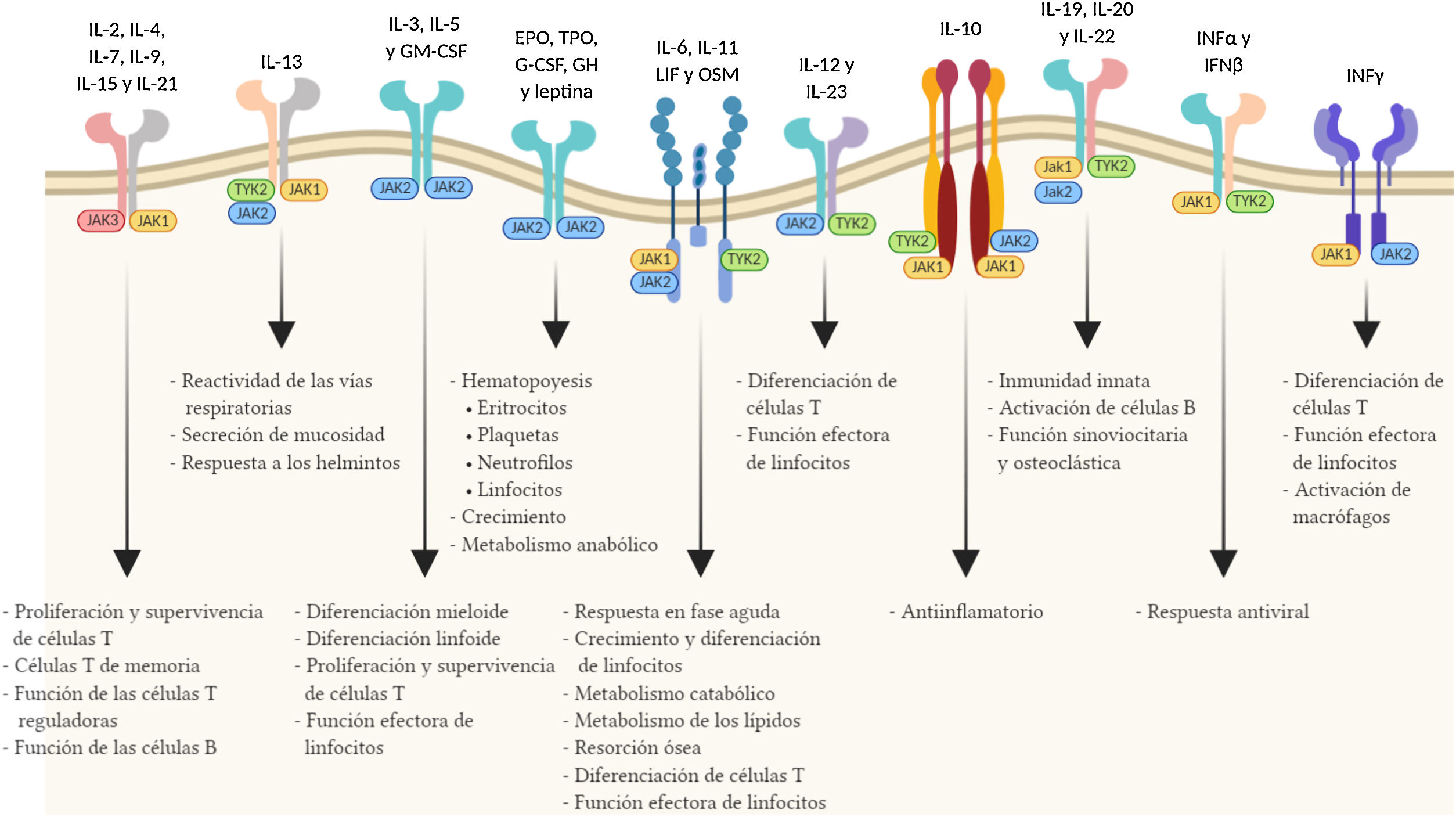
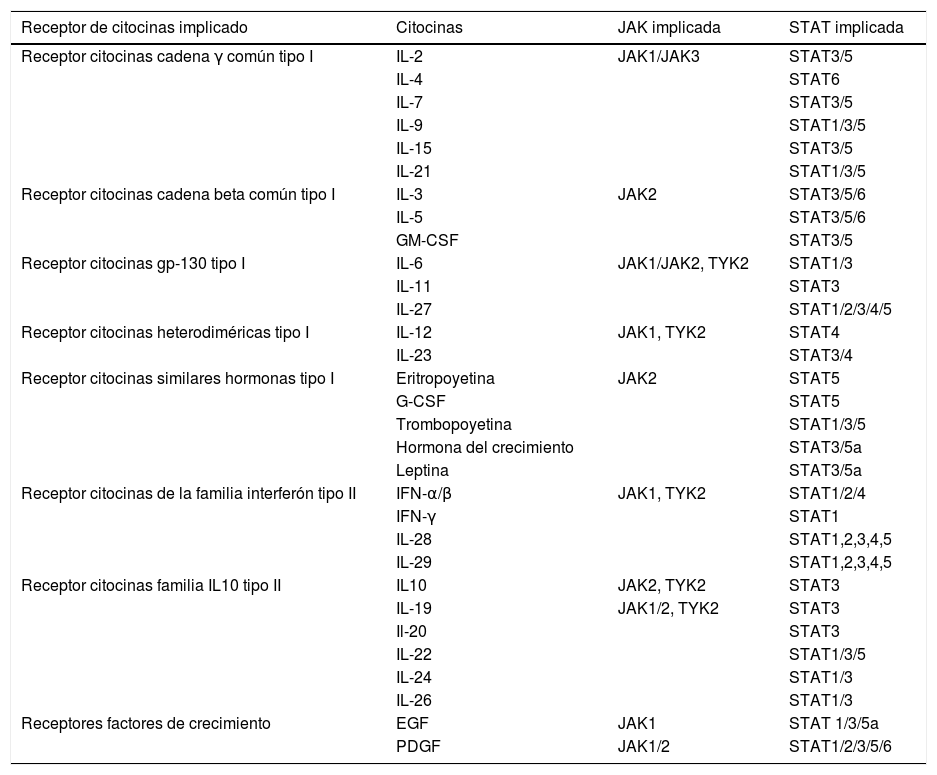
Mecanismo de acción de tofacitinibLas quinasas Janus están en el origen de diversas respuestas inmunológicas e inflamatorias y engloban 4 enzimas intracelulares de tipo tirosina-quinasa (JAK1, JAK2, JAK3 y TYK2 [tyrosine kinase-2]) asociadas con la región membranosa intracelular de distintos receptores que convierten las señales extracelulares, mediadas por diversas citocinas u hormonas, en procesos intracelulares11. La unión de una citocina al receptor causa su dimerización e induce la activación de las JAK asociadas. Cuando esto ocurre, las JAK fosforilan residuos específicos del dominio citoplasmático del receptor, donde se anclan los transductores de señales llamados signal transducers and activators of transcription (STAT). Entonces se produce la fosforilación de los STAT que una vez activados se dimerizan y traslocan al núcleo, donde participan en la expresión de múltiples proteínas12,13. Normalmente, las JAK actúan en parejas formadas por JAK distintas, aunque JAK2 se combina también consigo misma. Cada pareja de JAK es específica para un grupo diferente de citocinas (tabla 1), activando así distintas señales de transducción y, por lo tanto, respuestas determinadas ante los diversos estímulos14.
Vías de señalización JAK-STAT
| Receptor de citocinas implicado | Citocinas | JAK implicada | STAT implicada |
|---|---|---|---|
| Receptor citocinas cadena γ común tipo I | IL-2 | JAK1/JAK3 | STAT3/5 |
| IL-4 | STAT6 | ||
| IL-7 | STAT3/5 | ||
| IL-9 | STAT1/3/5 | ||
| IL-15 | STAT3/5 | ||
| IL-21 | STAT1/3/5 | ||
| Receptor citocinas cadena beta común tipo I | IL-3 | JAK2 | STAT3/5/6 |
| IL-5 | STAT3/5/6 | ||
| GM-CSF | STAT3/5 | ||
| Receptor citocinas gp-130 tipo I | IL-6 | JAK1/JAK2, TYK2 | STAT1/3 |
| IL-11 | STAT3 | ||
| IL-27 | STAT1/2/3/4/5 | ||
| Receptor citocinas heterodiméricas tipo I | IL-12 | JAK1, TYK2 | STAT4 |
| IL-23 | STAT3/4 | ||
| Receptor citocinas similares hormonas tipo I | Eritropoyetina | JAK2 | STAT5 |
| G-CSF | STAT5 | ||
| Trombopoyetina | STAT1/3/5 | ||
| Hormona del crecimiento | STAT3/5a | ||
| Leptina | STAT3/5a | ||
| Receptor citocinas de la familia interferón tipo II | IFN-α/β | JAK1, TYK2 | STAT1/2/4 |
| IFN-γ | STAT1 | ||
| IL-28 | STAT1,2,3,4,5 | ||
| IL-29 | STAT1,2,3,4,5 | ||
| Receptor citocinas familia IL10 tipo II | IL10 | JAK2, TYK2 | STAT3 |
| IL-19 | JAK1/2, TYK2 | STAT3 | |
| Il-20 | STAT3 | ||
| IL-22 | STAT1/3/5 | ||
| IL-24 | STAT1/3 | ||
| IL-26 | STAT1/3 | ||
| Receptores factores de crecimiento | EGF | JAK1 | STAT 1/3/5a |
| PDGF | JAK1/2 | STAT1/2/3/5/6 |
EGF: factor de crecimiento epidérmico; G-CSF: factor estimulante de colonias de granulocitos; GM-CSF: factor estimulante de colonias de granulocitos-macrófagos; IFN: interferón; IL: interleucina; JAK: quinasa Janus; PDGF: factor de crecimiento derivado de plaquetas; STAT: signal transducers and activators of transcription; TYK: tyrosine kinase.
La patogénesis de la CU es multifactorial, resultante probablemente de una respuesta inmunológica anómala a componentes de la microbiota en personas genéticamente susceptibles expuestas a desencadenantes ambientales. El equilibrio entre mediadores pro- y antiinflamatorios se ve alterado en estos casos. Por tanto, el bloqueo de la vía JAK/STAT, al interferir en las acciones de múltiples citocinas al mismo tiempo, podría modular de forma más amplia la respuesta inmunitaria innata y adaptativa y ser efectivo para frenar el proceso inflamatorio crónico que ocurre en la CU15,16.
Tofacitinib es un inhibidor potente de la familia de las JAK que interfiere con la señalización de JAK-STAT compitiendo con el ATP por la unión al dominio de la cinasa de JAK. Los estudios in vitro han demostrado una inhibición preferente de JAK1 y JAK3, con menos efecto sobre JAK217. Como resultado, tofacitinib inhibe la fosforilación y la activación de JAK, evitando la fosforilación y la activación de STAT y la posterior transcripción génica. Esto provoca que haya una menor producción de citocinas y una modulación de la respuesta inmunológica tanto innata como adaptativa18.
A consecuencia del papel de las vías JAK en el crecimiento, maduración y diferenciación celular y la hematopoyesis, es necesario considerar que su inhibición también se acompaña de un perfil de seguridad que pudiera condicionar su efectividad clínica en la CU (fig. 1).
Diferencias entre tofacitinib y los tratamientos biológicosDiferencias en el mecanismo de acción
Mientras que los fármacos biológicos inhiben profundamente y de forma prolongada la citocina diana correspondiente, los inhibidores de JAK modulan de forma más graduable y más reversible la respuesta inflamatoria15. Esta inhibición parcial y reversible de múltiples citocinas con tofacitinib da lugar a un perfil terapéutico diferenciado del de los fármacos biológicos.
Diferencias en sus propiedades farmacocinéticasEl hecho de que los medicamentos biológicos sean proteínas de gran tamaño y que tofacitinib sea una molécula sintética pequeña determina las grandes diferencias en su farmacocinética, con importantes repercusiones en su pauta de administración y su posología.
Absorción y distribución. Una de las ventajas de tofacitinib es su administración oral; resiste la degradación gástrica y puede entrar rápidamente en la circulación sistémica19. Tofacitinib se caracteriza por una buena absorción, con o sin alimentos (con una biodisponibilidad del 74%), un inicio rápido de su efecto (las concentraciones plasmáticas máximas se alcanzan de 30 a 60min) y un aumento de la exposición sistémica proporcional a la dosis (tabla 2). La estabilidad de concentraciones plasmáticas se alcanza en 24-48h, con una acumulación insignificante tras la posología de dos veces al día (2v/d)10. No parece existir influencia alguna de la edad, el sexo, el peso corporal (no requiere ajuste de dosis por peso) o la gravedad inicial de la enfermedad sobre la concentración plasmática promedio de tofacitinib20. Tofacitinib se une moderadamente a proteínas plasmáticas, sobre todo a la albúmina y, en individuos sanos, no afecta al filtrado glomerular, ni al flujo plasmático renal ni al aclaramiento de creatinina10,21.
Características farmacológicas de tofacitinib y de los medicamentos biológicos
| Medicamentos biológicos | Moléculas pequeñas | |
|---|---|---|
| Peso molecular | >1kDa | <700Da |
| Ruta de administración | Parenteral | Oral |
| Vida media | 9-25 días | 3h |
| Biodisponibilidad | 50-100% (según vía de administración) | 74% |
| Eliminación | Catabolismo proteico | Metabolismo hepático y excreción renal |
Los medicamentos biológicos se administran por vía parenteral (s.c. o i.v.). Su aclaramiento plasmático puede verse influido por diversos factores como son el peso, la albúmina sérica, la propia carga inflamatoria de la enfermedad o el desarrollo de anticuerpos contra el fármaco22. La distribución de los anticuerpos monoclonales en los tejidos está limitada por su gran tamaño e hidrofilia; por ejemplo, el tratamiento i.v. con infliximab induce de forma inmediata una alta concentración sistémica del fármaco. Sin embargo, con aquellos anticuerpos que son administrados s.c. la absorción se realiza por vía linfática y su concentración máxima se consigue varios días después.
Biotransformación y eliminación. La eliminación de tofacitinib es rápida (la vida media es de unas 3h) y los estudios en voluntarios sanos indican que el 95% del fármaco (hasta 100mg, dosis única) se elimina en 24h10. En caso de necesidad de suspensión por efectos adversos o situaciones como embarazo o cirugía, esto supone una ventaja, ya que su aclaramiento es rápido. Tofacitinib se elimina en un 70% por metabolismo hepático y en un 30% por excreción renal. Su metabolismo está mediado principalmente por CYP3A4, con una contribución menor de CYP2C1910. Es metabolizado en al menos 8 metabolitos, pero la actividad farmacológica se atribuye a la molécula de origen23. Los medicamentos biológicos se eliminan más lentamente por catabolismo proteico (la vida media de infliximab es de 9 a 12 días, la de adalimumab y golimumab es de 2 semanas y la de vedolizumab, de 25 días)22,24.
Interacciones farmacológicas. Los niveles circulantes de tofacitinib y su exposición aumentarán cuando se administre junto con inhibidores potentes del CYP3A4 (p.ej., ketoconazol, itraconazol, voriconazol, claritromicina) o cuando la administración de uno o más medicamentos de forma concomitante dé lugar a una inhibición moderada del CYP3A4 y la inhibición potente del CYP2C19 (p.ej., fluconazol). En estos casos, se recomienda disminuir la dosis de tofacitinib a la mitad. Por el contrario, los niveles circulantes de tofacitinib disminuirán cuando se administre junto con inductores potentes del CYP (p.ej., rifampicina), con la consiguiente reducción o incluso pérdida de respuesta por lo que no se recomienda su coadministración cuando se instaura un tratamiento con tofacitinib10. Debido al riesgo de inmunosupresión adicional, no se recomienda administrar azatioprina, tacrolimus o ciclosporina junto con tofacitinib9.
Situaciones especiales. No se ha establecido la seguridad y la eficacia de tofacitinib en menores de 18años. En pacientes de edad avanzada (65años o más), debido a un mayor riesgo de infecciones, tofacitinib únicamente se debe considerar si no hay un tratamiento alternativo adecuado disponible10. A diferencia de los anticuerpos monoclonales, la dosis de tofacitinib debe reducirse en pacientes con insuficiencia renal grave o hepática moderada, y está contraindicado en caso de insuficiencia hepática grave10.
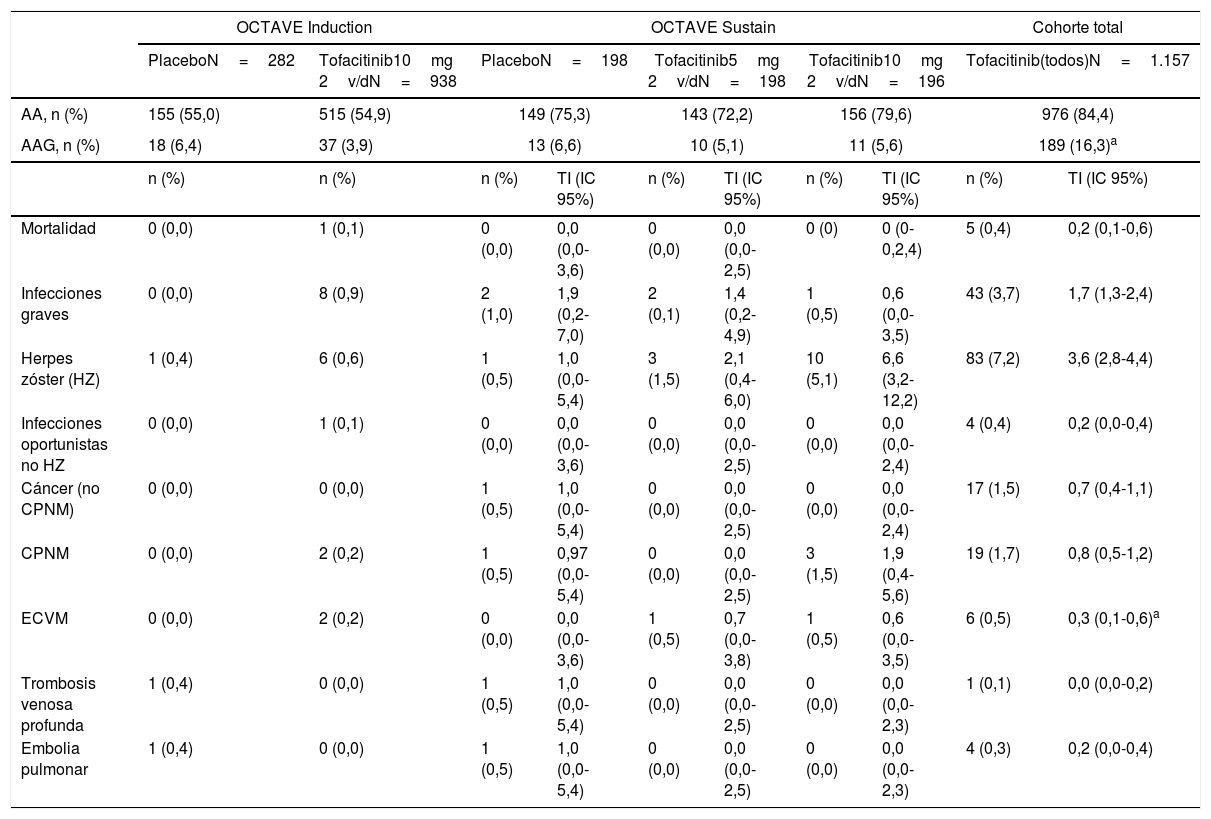
Seguridad de tofacitinib en el tratamiento de la colitis ulcerosaLa seguridad de tofacitinib en pacientes con CU ha sido estudiada en un ensayo clínico de fase 2 y en los estudios de fase 3 de inducción OCTAVE-induction 1 y OCTAVE-induction 2, de mantenimiento OCTAVE-Sustain y en el estudio de extensión OCTAVE-Open. Las cohortes analizadas son inducción (8 semanas), mantenimiento (52 semanas) y total (incluye cualquier paciente que recibiera tofacitinib 5mg o 10mg 2v/d en cualquiera de los estudios). La duración total del seguimiento con tofacitinib ha alcanzado los 6,1años, y se incluyeron 1.157 pacientes con una exposición de 2.403 pacientes-año. Durante el estudio de extensión, el 83% de los pacientes recibieron la dosis de 10mg 2v/d, una consideración importante teniendo en cuenta que alguno de los efectos adversos de tofacitinib son dosis-dependientes. Se ha constatado un perfil de seguridad general aceptable con ambas dosis20,25–27, y diversos metaanálisis avalan un perfil de seguridad de tofacitinib equiparable al de los fármacos biológicos28–30.
En las cohortes de inducción (OCTAVE-induction 1 y 2), la proporción de pacientes con acontecimientos adversos (AA), acontecimientos adversos graves (AAG) y suspensiones debidas a AA fueron similares en todos los grupos de tratamiento (tabla 3). En la cohorte de mantenimiento, la proporción de pacientes con AA y AAG fueron también comparables25. Los AA más frecuentes en pacientes que estaban tomando tofacitinib 10mg 2v/d en los estudios de inducción fueron cefalea, nasofaringitis, náuseas y artralgia. En los estudios de inducción y mantenimiento, en general, las categorías más frecuentes de AAG fueron los trastornos gastrointestinales y las infecciones, y el AAG más frecuente fue el empeoramiento de la CU10.
Resultados de seguridad durante el desarrollo clínico de tofacitinib en CU. Los datos de la cohorte general incluyen hasta 6,1años de seguimiento
| OCTAVE Induction | OCTAVE Sustain | Cohorte total | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PlaceboN=282 | Tofacitinib10mg 2v/dN=938 | PlaceboN=198 | Tofacitinib5mg 2v/dN=198 | Tofacitinib10mg 2v/dN=196 | Tofacitinib(todos)N=1.157 | |||||
| AA, n (%) | 155 (55,0) | 515 (54,9) | 149 (75,3) | 143 (72,2) | 156 (79,6) | 976 (84,4) | ||||
| AAG, n (%) | 18 (6,4) | 37 (3,9) | 13 (6,6) | 10 (5,1) | 11 (5,6) | 189 (16,3)a | ||||
| n (%) | n (%) | n (%) | TI (IC 95%) | n (%) | TI (IC 95%) | n (%) | TI (IC 95%) | n (%) | TI (IC 95%) | |
| Mortalidad | 0 (0,0) | 1 (0,1) | 0 (0,0) | 0,0 (0,0-3,6) | 0 (0,0) | 0,0 (0,0-2,5) | 0 (0) | 0 (0-0,2,4) | 5 (0,4) | 0,2 (0,1-0,6) |
| Infecciones graves | 0 (0,0) | 8 (0,9) | 2 (1,0) | 1,9 (0,2-7,0) | 2 (0,1) | 1,4 (0,2-4,9) | 1 (0,5) | 0,6 (0,0-3,5) | 43 (3,7) | 1,7 (1,3-2,4) |
| Herpes zóster (HZ) | 1 (0,4) | 6 (0,6) | 1 (0,5) | 1,0 (0,0-5,4) | 3 (1,5) | 2,1 (0,4-6,0) | 10 (5,1) | 6,6 (3,2-12,2) | 83 (7,2) | 3,6 (2,8-4,4) |
| Infecciones oportunistas no HZ | 0 (0,0) | 1 (0,1) | 0 (0,0) | 0,0 (0,0-3,6) | 0 (0,0) | 0,0 (0,0-2,5) | 0 (0,0) | 0,0 (0,0-2,4) | 4 (0,4) | 0,2 (0,0-0,4) |
| Cáncer (no CPNM) | 0 (0,0) | 0 (0,0) | 1 (0,5) | 1,0 (0,0-5,4) | 0 (0,0) | 0,0 (0,0-2,5) | 0 (0,0) | 0,0 (0,0-2,4) | 17 (1,5) | 0,7 (0,4-1,1) |
| CPNM | 0 (0,0) | 2 (0,2) | 1 (0,5) | 0,97 (0,0-5,4) | 0 (0,0) | 0,0 (0,0-2,5) | 3 (1,5) | 1,9 (0,4-5,6) | 19 (1,7) | 0,8 (0,5-1,2) |
| ECVM | 0 (0,0) | 2 (0,2) | 0 (0,0) | 0,0 (0,0-3,6) | 1 (0,5) | 0,7 (0,0-3,8) | 1 (0,5) | 0,6 (0,0-3,5) | 6 (0,5) | 0,3 (0,1-0,6)a |
| Trombosis venosa profunda | 1 (0,4) | 0 (0,0) | 1 (0,5) | 1,0 (0,0-5,4) | 0 (0,0) | 0,0 (0,0-2,5) | 0 (0,0) | 0,0 (0,0-2,3) | 1 (0,1) | 0,0 (0,0-0,2) |
| Embolia pulmonar | 1 (0,4) | 0 (0,0) | 1 (0,5) | 1,0 (0,0-5,4) | 0 (0,0) | 0,0 (0,0-2,5) | 0 (0,0) | 0,0 (0,0-2,3) | 4 (0,3) | 0,2 (0,0-0,4) |
AA: acontecimientos adversos; AAG: AA grave; CPNM: cáncer de piel no melanoma; CU: colitis ulcerosa; ECVM: evento cardiovascular mayor; IC: intervalo de confianza; TI: tasa de incidencia.
En cuanto a la mortalidad, ocurrieron 5 fallecimientos en toda la población tratada. Uno de ellos, que recibió tofacitinib 10mg 2v/d, murió por la rotura de un aneurisma aórtico durante el período de inducción, mientras que los otros 4 pacientes, que recibían tofacitinib 10mg 2v/d en el estudio de extensión abierto, fallecieron por angiosarcoma hepático, leucemia mieloide aguda, embolia pulmonar en el contexto de un colangiocarcinoma con metástasis peritoneal y melanoma, respectivamente. La tasa de incidencia (TI, expresada como pacientes con ≥1 evento por 100 pacientes-año de exposición) de muerte fue de 0,2 (intervalo de confianza [IC] del 95% 0,1-0,6)26.
Infecciones, herpes zóster y tuberculosisInfecciones. En los ensayos OCTAVE-induction 1 y 2, las infecciones graves fueron más frecuentes con tofacitinib (0,9%) que con placebo (0%). En el OCTAVE-Sustain, las TI de infección grave fueron similares en los 3 grupos de tratamiento20. En la cohorte total hasta 6,1años tratada con tofacitinib, la TI de infección grave fue 1,75 (IC 95% 1,27-2,36)27 (tabla 3). Ningún caso de infección grave en el programa de desarrollo clínico en CU tuvo un desenlace fatal. La edad avanzada se ha asociado con mayor riesgo de infecciones oportunistas y herpes zóster (HZ), pero no con las infecciones graves31. El comité de seguridad de la EMA, tras la reevaluación de los datos procedentes del estudio clínico en marcha (A3921133) en pacientes con AR de 50años o mayores y al menos un factor de riesgo cardiovascular, así como los resultados de estudios previos en todas las indicaciones, ha observado un riesgo aumentado de infecciones graves y mortales en pacientes de 65años o más32.
Herpes zóster. En la cohorte de mantenimiento, se ha observado una mayor TI (≥1 evento por 100 pacientes-año de exposición) de infección por HZ en los pacientes tratados con 5mg 2v/d (2,1) y con 10mg 2v/d (6,6) de tofacitinib que en el grupo placebo (1,0), siendo la diferencia estadísticamente significativa para la dosis de 10mg 2v/d (tabla 3). Es decir, que el tratamiento con tofacitinib se asocia con un riesgo de HZ de forma dosis-dependiente. En la cohorte total hasta 6,1años, la TI fue 3,57 casos 100 pacientes-año27. En el análisis de seguridad de hasta 4,4años de duración, muchos de los casos fueron clasificados como leves o moderados (96%), y solo hubo 6 casos de HZ diseminado y 3 casos de neuralgia posherpética33. Un 85% de los casos se resolvieron con un tratamiento antiviral (hasta el momento del análisis) y en la mayoría de las ocasiones sin necesidad de retirar tofacitinib (44 de 65 casos); se decidió interrumpir temporalmente la dosis de tofacitinib en 16 pacientes. Los factores de riesgo independientes para la infección por HZ fueron la edad (p<0,0001) y el fallo previo a iTNF (p<0,05), pero no la duración del tiempo en tratamiento con tofacitinib33.
Infecciones oportunistas. Otras infecciones oportunistas fueron infrecuentes en los pacientes con CU tratados con tofacitinib (tabla 3). La TI (excluyendo HZ) en la cohorte total de 1.157 pacientes fue de 0,16 (IC 95% 0,04-0,42), sin evidencia de aumento del riesgo de infección oportunista con la duración del tratamiento27. Los 4 casos ocurridos consistieron en colitis por citomegalovirus, criptococosis pulmonar, histoplasmosis y hepatitis por citomegalovirus26,27.
Aunque en los ensayos clínicos de inducción y mantenimiento con tofacitinib en CU no se ha registrado ningún caso de tuberculosis20, se recomienda seguir las directrices locales respecto a la detección de tuberculosis latente10.
VacunaciónLos pacientes con EII presentan un mayor riesgo de infecciones graves y HZ que las personas sin esta enfermedad; además, el tratamiento con tofacitinib, pero también con biológicos, tiopurinas, corticosteriodes y terapias combinadas, aumentan este riesgo34–36. Por tanto, parece recomendable valorar la necesidad de vacunar a estos pacientes como medida preventiva37. Además, antes de iniciar el tratamiento con tofacitinib es recomendable la puesta al día del calendario vacunal, tal y como indican las guías clínicas38. No se recomienda administrar vacunas de microorganismos vivos simultáneamente con tofacitinib. La inmunización con vacunas de microorganismos vivos debe realizarse al menos 2 semanas, aunque es preferible 4 semanas, antes del inicio del tratamiento con tofacitinib10.
La EMA ha autorizado una vacuna inactivada para HZ de virus recombinante (Shingrix®), abriendo la posibilidad de evitar otras vacunas con virus atenuados en estos pacientes susceptibles. Aunque actualmente no hay recomendaciones definitivas sobre la vacunación frente al HZ en Europa, el Grupo Español de Trabajo en Enfermedad de Crohn y Colitis ulcerosa recomienda el uso de Shingrix® para pacientes que reciben tratamiento inmunosupresor en su documento sobre vacunas y algoritmos de actuación en pacientes con EII39. El Advisory Committee on Immunization Practice recomienda la vacunación con la vacuna recombinante en todos los adultos mayores de 50años y en pacientes con antecedentes de HZ, enfermedades crónicas y pacientes en inmunosupresión: estos dos últimos grupos incluirían a la gran mayoría de los pacientes con EII40.
NeoplasiasEn la cohorte total del análisis integrado de seguridad (hasta 6,1años de seguimiento), la TI de neoplasias (excluyendo el cáncer de piel no melanoma [CPNM]) fue 0,69 (IC 95% 0,4-1,11) (tabla 3). Dado que una proporción de estos pacientes había recibido también otros inmunosupresores como iTNF o tiopurinas, la causalidad de estos tumores podría ser debatible. Hubo 17 casos de cáncer (2 casos de cáncer de cuello uterino, 2 de cáncer de mama y 2 de adenocarcinoma de colon; uno de angiosarcoma hepático, uno de colangiocarcinoma, uno de leiomiosarcoma cutáneo, uno de linfoma difuso de células B grandes, uno de linfoma asociado al virus de Epstein-Barr, uno de melanoma, uno de carcinoma de células renales, uno de cáncer de pulmón, uno de carcinoma de mama ductal invasivo, uno de trombocitemia esencial, uno de leucemia mieloide aguda, más neoplasias secundarias en el hígado y el peritoneo41.
La TI de CPNM fue 0,78 (IC 95% 0,47-1,22) (tabla 3); en el análisis hasta 6,1años, de los 19 pacientes con CPNM, 17 habían estado expuestos a tiopurinas y 15 a iTNF. Estas TI son similares a las de pacientes con CU tratados con medicamentos biológicos41,42.
Lípidos y complicaciones cardiovascularesSe observó un incremento de la concentración de lípidos en sangre (colesterol total [CT], LDLc y HDLc) en los pacientes tratados con tofacitinib20. Los cambios en el perfil lipídico se estabilizaron después de 4-8 semanas de tratamiento y revirtieron al abandonarlo. Los cocientes CT/HDLc y LDLc/HDLc, que son mejores parámetros predictivos de riesgo cardiovascular que los lípidos individuales, permanecieron sin cambios43. En total, a lo largo de los 4,4años de seguimiento con tofacitinib se prescribió un nuevo tratamiento hipolipemiante y se incrementó la dosis de hipolipemiantes al 4,8% y 1,5%, respectivamente, de la población del estudio43. Se recomienda examinar las concentraciones de lípidos a las 4-8 semanas del comienzo del tratamiento10 (tabla 4).
Recomendaciones para el control clínico de los pacientes tratados con tofacitinib
| Al inicio | 4-8 semanas tras el inicio | Cada 3 meses a partir de entonces | |
|---|---|---|---|
| Linfocitos | |||
| Neutrófilos | |||
| Hemoglobina | |||
| Lípidos | |||
| Enzimas hepáticas | También se recomienda controlar periódicamente los valores hepáticos e investigar inmediatamente el motivo de toda elevación de las enzimas hepáticas | ||
| Evitar iniciar el tratamiento en pacientes con: | Recuento de linfocitos<750células/mm3 | Recuento de neutrófilos<1.000células/mm3 | Niveles de hemoglobina<9g/dl |

Creado a partir de la ficha técnica de Xeljanz®.
Las complicaciones cardiovasculares graves fueron infrecuentes en los ensayos de tofacitinib en pacientes con CU (TI 0,3; IC 95% 0,1-0,6)26. En la cohorte total hasta 4,4años hubo AA cardiovasculares confirmados (evento cardiovascular mayor [ECVM]) en 4 pacientes que recibieron tofacitinib: un ictus hemorrágico, una disección aórtica, un síndrome coronario agudo y un infarto agudo de miocardio25. Tres de los 4 pacientes tenían 4 o más factores de riesgo basales, incluyendo hiperlipidemia43.
Eventos tromboembólicosLos pacientes con enfermedades inflamatorias tienen un riesgo incrementado de trombosis venosa profunda (TVP) y embolia pulmonar (EP)44. En CU las TI son 0,07-0,30 para TVP y 0,04-0,20 para EP45. En la cohorte total del programa de desarrollo clínico de tofacitinib en CU (≤6,1años de seguimiento), un paciente tuvo una TVP (TI 0,04; IC 95% 0,00-0,23) y 4 experimentaron EP (TI 0,16; IC 95% 0,04-0,41); la dosis de tofacitinib predominante fue 10mg 2v/d; todos presentaban factores de riesgo de tromboembolismo45.
Tofacitinib puede elevar el de riesgo de tromboembolismo venoso (TEV), tanto de TVP como de EP, en pacientes de alto riesgo (TEV previo, cirugía mayor, inmovilización, infarto de miocardio en los 3 meses anteriores, insuficiencia cardíaca, uso de anticonceptivos hormonales combinados o terapia hormonal sustitutiva, trastorno hereditario de la coagulación y neoplasia maligna; otros factores de riesgo adicionales de TEV son edad avanzada, obesidad, diabetes, hipertensión y tabaquismo)32. Independientemente de la indicación y la dosis, tofacitinib debe utilizarse con precaución en todos los pacientes con factores de riesgo de TEV.
La evaluación de los datos apunta a que el riesgo de TEV es mayor en los pacientes que toman tofacitinib 10mg 2v/d y en aquellos que son tratados durante un período prolongado. Por ello, no se recomiendan dosis de mantenimiento de 10mg 2v/d en pacientes con CU con factores de riesgo de TEV, a menos que no haya un tratamiento alternativo adecuado10,32,46. Se recomienda en todos los pacientes tratados con tofacitinib realizar un seguimiento para detectar signos y síntomas sugestivos de TEV, instruyéndoles para que soliciten atención médica de inmediato si experimentan dichos síntomas32,46.
Pruebas analíticasDurante los estudios de inducción, los niveles de neutrófilos, linfocitos y hemoglobina permanecieron estables. En el estudio de mantenimiento, las TI de anemia fueron 2,91, 5,51 y 2,55 para los grupos de placebo, tofacitinib 5mg 2v/d y tofacitinib 10mg 2v/d, respectivamente. Las TI de neutropenia fueron 0, 0,67 y 0,64, respectivamente, mientras que no se notificaron linfopenias47. No se han observado cambios importantes en los valores de laboratorio desde el inicio de la fase de extensión abierta hasta los 36 meses de seguimiento, con excepción de los lípidos48. En cualquier caso, la monitorización del hemograma es indispensable siguiendo las recomendaciones mostradas en la tabla 4.
GestaciónEstá contraindicado utilizar tofacitinib durante el embarazo y la lactancia10. Sin embargo, se han dado algunos casos de exposición a tofacitinib de madres (n=74) o padres (n=84) en el momento de la concepción o antes o durante el embarazo en los estudios con tofacitinib para todas las indicaciones49. En aquellos con CU, se han identificado 11 casos de exposición materna y 14 casos de exposición paterna a tofacitinib. Los resultados incluyeron 15 recién nacidos sanos, ninguna muerte fetal, ninguna muerte neonatal, ninguna malformación congénita, 2 abortos espontáneos y 2 abortos inducidos. A pesar de que los datos son escasos, estos resultados parecen similares a los publicados sobre otras poblaciones expuestas a tofacitinib y a los de la población general49.
Seguridad de tofacitinib en pacientes con artritis reumatoideTofacitinib fue primeramente evaluado y autorizado para el tratamiento de la AR, por lo que existen datos de su seguridad a más largo plazo que en la indicación de CU. En el análisis integrado de seguridad de tofacitinib en 7.061 pacientes con AR (22.875 pacientes-año de exposición) de hasta 9,5años y con una mediana de exposición de 3,1años y un 30% de pacientes con más de 5años de exposición, la TI (expresada como pacientes con ≥1 evento por 100 pacientes-año de exposición) de AAG fue 9,0 (IC 95% 8,6-9,4)50. Hubo infecciones graves en 576 pacientes (8,2%; TI 2,5; IC 95% 2,3-2,7) y 782 pacientes (11,1%) desarrollaron HZ (TI 3,6; IC 95% 3,4-3,9). Las neoplasias (excluyendo CPNM) se produjeron en 177 pacientes (2,5%; TI 0,8; IC 95% 0,7-0,9), CPNM en 129 pacientes (1,8%; TI 0,6; IC 95% 0,5-0,7) y linfomas en 12 pacientes (0,2%; TI 0,1; IC 95% 0,0-0,1). Un total de 85 pacientes (1,3%) experimentaron ECVM (TI 0,4; IC 95% 0,3-0,5), 27 (0,4%) tuvieron TEV (TI 0,12; IC 95% 0,08-0,17) y 28 (0,4%) tuvieron EP (TI 0,12; IC 95% 0,08-0,17). Hubo 59 muertes (0,8%; TI 0,3; IC 95% 0,2-0,3)50.
El estudio ORAL Surveillance (A3921133) es un estudio postautorización de seguridad que compara tofacitinib frente iTNF, en curso y de gran tamaño muestral (n=4.362), en pacientes con AR de 50años de edad y mayores y con al menos un factor de riesgo cardiovascular. Los pacientes fueron aleatorizados a recibir tofacitinib 10mg 2v/d, tofacitinib 5mg 2v/d o un iTNF (etanercept 50mg una vez a la semana o adalimumab 40mg cada 2 semanas). La TI de EP fue estadísticamente superior en pacientes tratados con tofacitinib 10mg 2v/d (TI 0,54; IC 95% 0,32-0,87) en comparación con la de los iTNF (TI 0,09; IC 95% 0,02-0,26). La TI para la dosis de 5mg 2v/d (0,27; IC 95% 0,12-0,52) fue más alta en comparación con la de los iTNF, pero la diferencia no fue estadísticamente significativa32,51. El riesgo de infecciones graves (mortales y no mortales) aumentó más en pacientes mayores de 65años, en comparación con los pacientes más jóvenes10. Respecto a la mortalidad, no hubo diferencias estadísticamente significativas con la dosis de 5mg 2v/d pero sí entre la dosis de 10mg 2v/d (TI 0,89; IC 95% 0,59-1,29) y los iTNF (TI 0,27; IC 95% 0,12-0,51)32,51.
ConclusiónLa seguridad de tofacitinib ha sido estudiada en pacientes con CU de moderada a grave, en el tratamiento de inducción y de mantenimiento, con un seguimiento en los ensayos clínicos de hasta 6,1años. El perfil de seguridad observado es aceptable y manejable, aunque son necesarios datos adicionales de práctica clínica para confirmarlo.
A la hora de prescribir tofacitinib es preciso valorar, por un lado, que los efectos adversos significativos aparecen durante la mayoría de los tratamientos de la CU, y por otra parte, tener en cuenta las ventajas que tofacitinib ofrece, tales como su administración oral, ausencia de inmunogenicidad y su vida media corta.
Conflicto de interesesAntonio López-Sanromán ha sido conferenciante para Pfizer y ha participado en sesiones de discusión a propósito de tofacitinib y de colitis ulcerosa; Juan V. Esplugues ha recibido honorarios por la realización de conferencias o consejo de experto y ayudas para la realización de proyectos de investigación, de las siguientes empresas: MSD, AbbVie, AstraZeneca, Gilead y Pfizer; Eugeni Domènech ha recibido honorarios por la realización de conferencias o consejo de experto, se le ha facilitado la asistencia a cursos o congresos, o su unidad ha recibido donaciones o ayudas para la realización de eventos o para proyectos de investigación, de las siguientes empresas: MSD, AbbVie, Takeda, Kern Pharma, Pfizer, Janssen, Celgene, Otsuka Pharmaceuticals, Adacyte, Ferring, Shire Pharmaceuticals, Tillots, Thermofisher, Grifols, Gebro.
Los autores desean agradecer a Mónica Valderrama, Ana Cábez y Susana Gómez de Pfizer la búsqueda bibliográfica y la revisión de este manuscrito, y a la Dra. Ana Moreno Cerro y a Anabel Herrero, PhD, la asistencia en la redacción realizada en nombre de Springer Healthcare y financiada por Pfizer España.

