







INTRODUCCIÓN
Los síndromes de poliposis gastrointestinales se caracterizan por la presencia de múltiples pólipos en el tubo digestivo, con una afección preferente del colon y el recto. Son un grupo de enfermedades de escasa incidencia, pero con muy variadas características, que precisan una correcta individualización con el fin de que pueda proporcionarse el tratamiento más adecuado. La clasificación más útil es la que combina la naturaleza hereditaria o no del proceso, con su histoenfermedad (adenoma, hamartoma, hiperplasia o proliferación linfoide) (tabla I)1. Otra forma práctica de clasificar los síndromes de poliposis gastrointestinal es según su riego de desarrollar cáncer colorrectal1.

La poliposis adenomatosa familiar (PAF) es la forma de poliposis más conocida y más frecuente. Se trata de una enfermedad hereditaria autosómica dominante con una prevalencia de aproximadamente 1 de cada 10.000-20.000 individuos2,3. La característica fundamental de la PAF es la aparición de más de 100 pólipos adenomatosos en el colon y el recto (pueden llegar a ser miles), iniciándose a una edad muy joven y con un riesgo de cáncer colorrectal cercano al 100% si el paciente no recibe tratamiento de forma precoz4,5. De todos los diagnósticos de cáncer colorrectal, los pacientes con PAF suponen únicamente menos de un 0,5%, pero la posibilidad de diagnóstico precoz y posterior tratamiento ha situado esta enfermedad en un punto de máximo interés científico4. Además, al tratarse de un trastorno o enfermedad hereditaria compleja, junto a la enfermedad colorrectal, pueden asociarse una gran cantidad de alteraciones en otros órganos que es preciso investigar antes del tratamiento y durante el seguimiento, o incluso pueden servir para el diagnóstico de PAF6. El objetivo principal de este artículo es hacer una revisión del estado actual en el manejo de los individuos afectados de PAF. Para ello, hemos revisado los artículos publicados más relevantes realizando previamente una búsqueda bibliográfica en la base de datos Medline, para identificar los referidos al diagnóstico, el tratamiento y el seguimiento de esta enfermedad.
MANIFESTACIONES CLÍNICAS
Historia natural de la enfermedad
La manifestación más prominente de la PAF es la aparición de múltiples pólipos adenomatosos en el colon y el recto, de manera que ante la sospecha clínica de esta enfermedad hay que efectuar una endoscopia digestiva baja. Hay que recordar que en un individuo afectado de PAF y sin tratamiento, las lesiones suelen desarrollarse en la segunda y la tercera décadas de la vida: el 15% a los 10 años de edad, el 50% de los pacientes sobre los 15 años y un 95% a los 35 años7. Por todo ello, se considera que, en el contexto de una familia diagnosticada de PAF, si un individuo no ha desarrollado pólipos antes de los 40 años, es muy improbable que presente manifestaciones clínicas de esta enfermedad8.
Clínica
Durante años la clínica puede ser silente, y suele consistir en deposiciones más blandas, a veces diarreicas y, ocasionalmente, dolor abdominal tipo cólico, sin que se afecte el estado general del paciente. La aparición de rectorragia suele ser un signo de alarma para los pacientes, y un porcentaje superior al 30% de los que consultan puede presentar ya malignización de uno o varios pólipos9. Sin tratamiento, la edad media de detección del cáncer es de 35 años10. En los casos no tratados la muerte suele acontecer a los 40-45 años, es decir, 20 años antes de la edad de fallecimiento por cáncer colorrectal esporádico9.
Aunque en la bibliografía aparezcan términos a veces confusos, como formas agresivas o graves de la PAF, referidas a la presentación con un número extremadamente elevado de pólipos y que se utiliza para indicar tratamientos con resecciones quirúrgicas más amplias, desde un punto de vista práctico es más útil diferenciar a los pacientes que tienen un número superior o inferior a 20 pólipos en el recto, o más o menos 1.000 pólipos en el colon1.
Un dato que vale la pena destacar es que aproximadamente un 20-30% de los pacientes con PAF no presenta historia familiar de la enfermedad, lo que significa que son casos de novo11. En estos casos, los padres y hermanos del paciente no presentarán la enfermedad, pero cada uno de los hijos tendrán un 50% de posibilidades de heredar la enfermedad (si la herencia es autosómica dominante), por lo que es completamente necesario realizar el consejo genético.
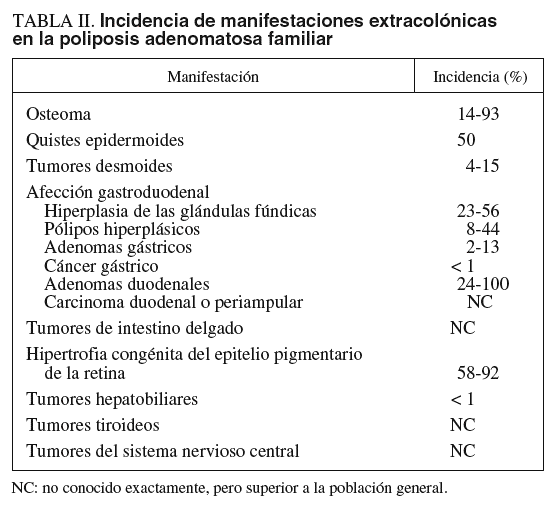
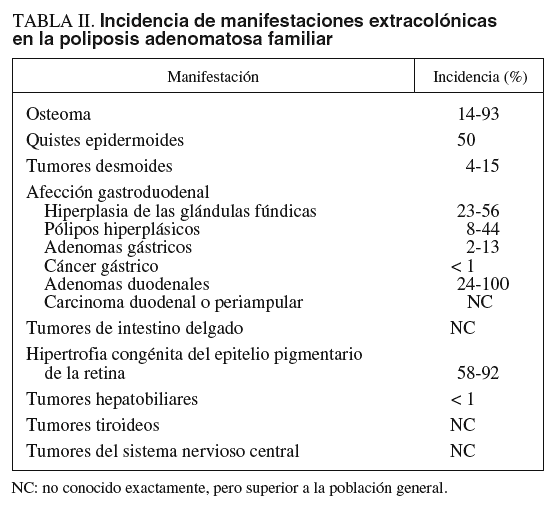
MANIFESTACIONES EXTRACOLÓNICAS
Las manifestaciones extracolónicas de la PAF (que pueden presentarse hasta en un 40% de los casos) configuran no sólo algunos síndromes individualizados, como el síndrome de Gardner (afección gastroduodenal, tumores desmoides y osteomas entre otras) o el síndrome de Turcot-Despres (tumores del sistema nervioso central), sino otras enfermedades referidas con elevada frecuencia en la PAF (tabla II)12,13. Se considera que estas manifestaciones extracolónicas son secundarias a una expresión variable de un único defecto genético más que el resultado de múltiples defectos genéticos diferentes8,10.
Lesiones en el tracto digestivo superior
Los pólipos de las glándulas fúndicas son los pólipos gástricos más frecuentes en la PAF3. Suelen estar presentes en el 50% de los pacientes, y normalmente se localizan en el fundus y en el cuerpo gástrico, extendiéndose en su distribución hacia el antro14. Histopatológicamente, se caracterizan por una dilatación y un cambio quístico de las glándulas fúndicas. Estos pólipos pueden aumentar en número, pero la probabilidad de transformación maligna es casi nula, aunque se ha descrito algunos casos de displasia15,16.
Los pólipos hiperplásicos son también frecuentes (el 8-44% de los pacientes) y tampoco se ha descrito malignización de éstos.
Curiosamente, los adenomas gástricos son los pólipos menos frecuentes en los pacientes con PAF (con una incidencia aproximada del 6%) y pueden presentarse de forma difusa por todas las áreas del estómago. Aunque éstos no suelen aumentar de tamaño ni variar su grado de atipia, de forma ocasional pueden trasformarse en malignos.
Si bien se ha descrito un aumento del riesgo de cáncer gástrico en series japonesas14, en los países occidentales el riesgo de presentar cáncer gástrico en los pacientes afectados de PAF se considera que no es significativamente superior al riesgo de la población general17.
En el intestino delgado, los adenomas asientan principalmente en el duodeno, cercanos a la papila duodenal (ampolla de Vater). Se encuentran en más del 80% de los pacientes con PAF, con un riesgo acumulado cercano al 100%18,19. Los pólipos duodenales normalmente son múltiples y sésiles, y se localizan predominantemente en los pliegues mucosos de la segunda porción duodenal, sobre todo en la región periampular. En ocasiones, la mucosa que aparece microscópicamente normal puede presentar cambios adenomatosos en el estudio histológico, aspecto que será importante en el seguimiento de estas lesiones20.
El potencial maligno de los adenomas sésiles duodenales es bajo. Sin embargo, la presencia de displasia en la región periampular es elevada y oscila entre el 60 y el 70%20. Para el manejo de los pólipos duodenales, éstos pueden clasificarse en 5 estadios (0-IV), según la clasificación propuesta por Spigelman et al21 y modificada posteriormente en la reunión de consenso de Viena según el grado de displasia22. Esta clasificación tiene en cuenta 4 variables: tamaño del pólipo, número de pólipos, grado de displasia y arquitectura del pólipo. El grado de displasia aumenta cuanto mayor es el estadio de Spigelman. Los pacientes con un estadio IV de Spigelman son los que presentan un mayor riesgo de carcinoma periampular, que se presenta en el 4% de los pacientes afectados de PAF, y es la causa de muerte más frecuente en los pacientes colectomizados18,19,21,23.
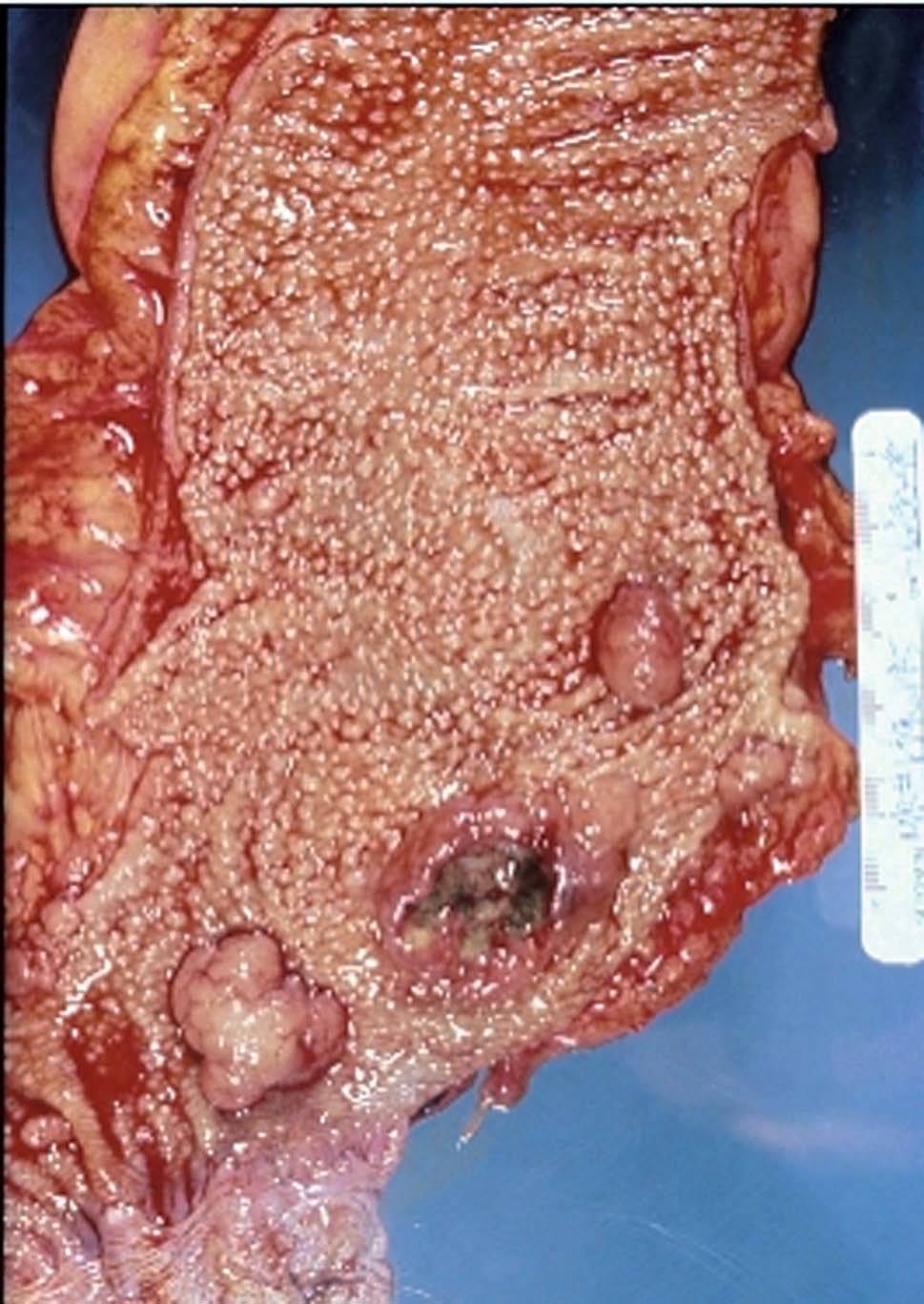
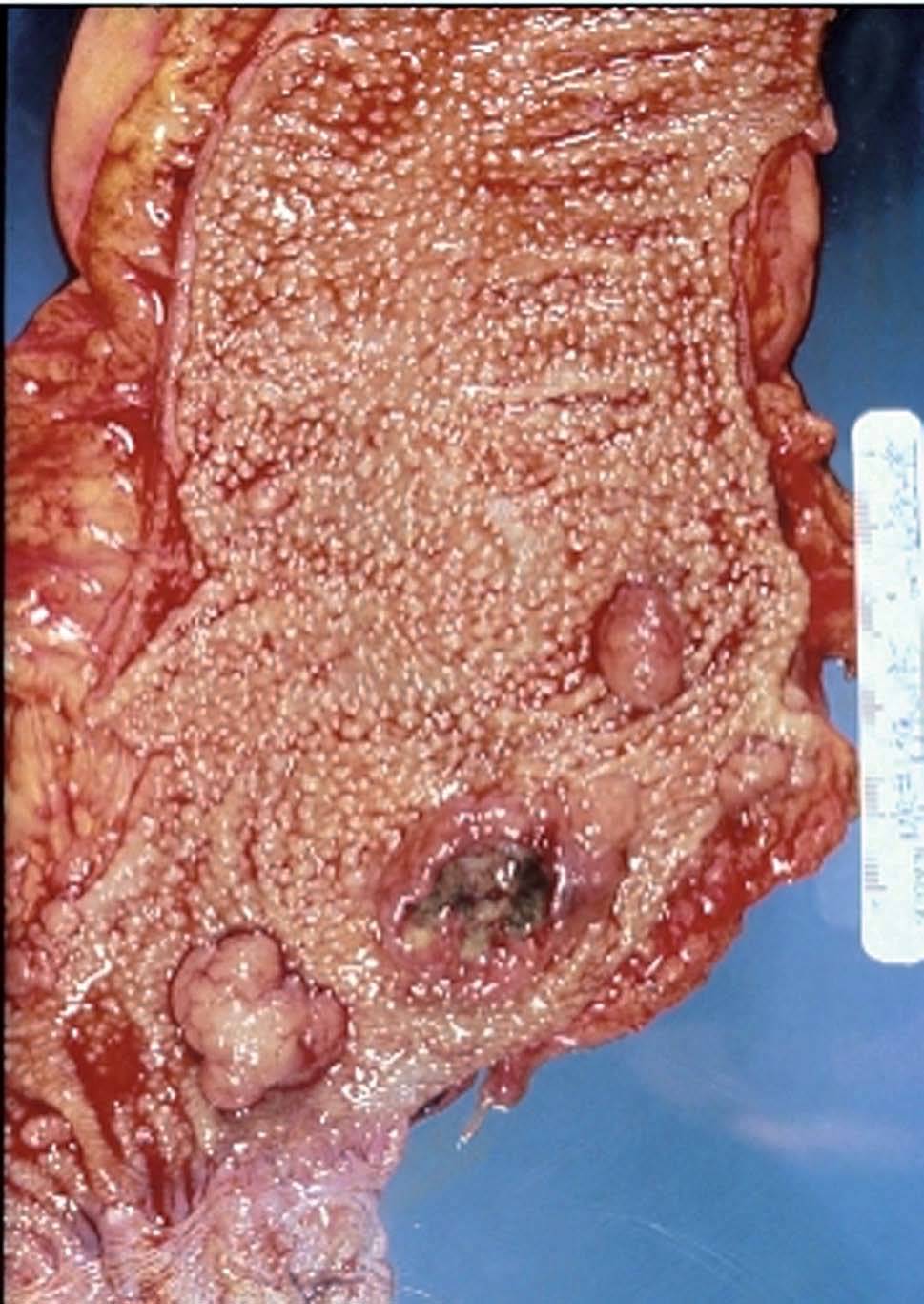
Tumores desmoides
Los tumores desmoides son histológicamente benignos, ya que en realidad se trata de un sobrecrecimiento de tejido fibroaponeurótico. Estas lesiones ocurren raramente en la población general, pero su incidencia en pacientes con PAF puede alcanzar el 15%24. Los tumores desmoides que afectan a estos pacientes son generalmente intraabdominales (fig. 1), menos frecuentes en la pared abdominal o las extremidades, y aparecen en la mayoría de casos durante el seguimiento en los primeros 3 años de cualquier intervención quirúrgica que se realice a estos pacientes24.

Fig.1. Imagen de tomografía computarizada abdominal que muestra la presencia de un gran tumor desmoide intraabdominal en un paciente afectado de poliposis adenomatosa familiar.
Tener una historia familiar de tumores desmoides es un factor de riesgo para el su desarrollo, y se asocian también con determinadas mutaciones específicas en el gen APC (adenomatous polyposis coli gene)25,26. Las mutaciones situadas entre los codones 1310 y 2011, por ejemplo, se asocian con un riesgo 6 veces mayor de desarrollar tumores desmoides, comparado con el riesgo descrito en las mutaciones situadas entre otras localizaciones. Por otro lado, hay familias con mutaciones en las que la manifestación de la PAF consiste en tumores desmoides con ausencia o presencia de muy pocos pólipos en el área colorrectal27,28. Recientemente, Sturt et al29 han sugerido que debe haber otros factores genéticos, diferentes del gen APC, implicados en la aparición de los tumores desmoides en la PAF.
Los tumores desmoides intraabdominales tienen tendencia a englobar el mesenterio del intestino delgado, con o sin afección de sus vasos principales24. La evolución de este tipo de tumores en esta localización varía ampliamente. Hay casos de enfermedad estable en pacientes asintomáticos y, contrariamente, casos de crecimiento agresivo de éstos. Un aspecto determinante en la falta de consenso en el tratamiento de los mismos es la ausencia de factores predictivos de respuesta o recidiva a cualquiera de los tratamientos descritos30. De hecho, junto al tratamiento quirúrgico se han descrito múltiples tratamientos médicos que abarcan el uso de antiinflamatorios, antiestrógenos, quimioterapia e incluso radioterapia en casos excepcionales26. Su resección quirúrgica, por la localización mesentérica, muchas veces es incompleta y está asociada con una alta morbilidad y tendencia a la recidiva27. Por ello, se recomienda tratar este tipo de tumores en función de su presentación y tendencia al crecimiento24. Sólo en casos excepcionales, en que haya complicaciones por la presencia del tumor desmoide y la resección no conlleve una elevada morbilidad, cabe considerar la intervención quirúrgica. Por el contrario, la resección de los tumores localizados en la pared abdominal es más fácil y se asocia con tasas bajas de recidiva local si el margen de resección es mayor de 1 cm24.
Osteomas
Los osteomas fueron descritos por primera vez por Gardner y Richards en 195313 y su incidencia estimada varía entre el 14 y el 93%. Estas lesiones pueden presentarse en cualquier hueso, aunque son mucho más frecuentes en el esqueleto facial, principalmente en el ángulo de la mandíbula, y en los huesos largos12,31. Estos tumores son benignos, pero pueden ocasionar síntomas debido a su crecimiento local. Los osteomas mandibulares pueden ser un buen marcador de manifestaciones extracolónicas de la PAF, en ausencia de un estudio genético o cuando éste no pone de manifiesto la presencia de esta enfermedad31. De esta manera, en una familia con la enfermedad, la aparición de un osteoma en un descendiente indica la transmisión hereditaria de ésta.
Quistes epidermoides
Los quistes epidermoides son lesiones benignas del tejido celular subcutáneo que suelen desarrollarse durante la pubertad32. En los pacientes con PAF pueden localizarse en las extremidades, la cara o el cuero cabelludo, mientras que en la población general suelen presentarse en la espalda. Lo más interesante de estas lesiones es que durante la infancia de los individuos de la población general son extraordinariamente infrecuentes, pero en la PAF pueden aparecer antes que los pólipos colorrectales8.
Hipertrofia congénita del epitelio pigmentario de la retina
Se trata de lesiones pigmentarias oscuras, redondas u ovales, pequeñas, con frecuencia periféricas que se detectan mediante retinoscopia. Estas lesiones son completamente asintomáticas. El estudio histológico de estas lesiones ha demostrado que en realidad se trata de hamartomas del epitelio pigmentario; sin embargo, el término más aceptado es el de hipertrofia congénita del epitelio pigmentario de la retina (HCEPR)33.
La prevalencia de la HCEPR en la PAF es muy variable (del 0 al 42%). Una de las explicaciones de dicha variabilidad es la distinta interpretación de las lesiones por parte de diferentes especialistas. Del mismo modo, es importante recordar que la HCEPR puede encontrarse hasta en el 2% de la población general34. A pesar de esta variabilidad y de la subjetividad en la interpretación, se ha propuesto utilizar la HCEPR como un marcador biológico de la enfermedad para identificar a los portadores asintomáticos de la PAF.
Lesiones malignas extracolónicas
Se han descrito otros tumores asociados con mayor frecuencia a la PAF respecto a la población general, como hepatoblastoma, o tumores tiroideos, adrenales, del árbol biliar, el páncreas y el sistema nervioso central23,35,36 (tabla II).
POLIPOSIS ADENOMATOSA FAMILIAR ATENUADA
Hay una forma atenuada de PAF que difiere de la forma clásica básicamente en la presencia de un número menor de pólipos (menos de 100) que tienen tendencia a desarrollarse en el colon derecho, y cuya presentación, tanto por lo que respecta a los pólipos cómo al cáncer, suele retrasarse unos 15 años en relación con la PAF clásica37,38. Habitualmente estos pacientes con poliposis adenomatosa familiar atenuada (PAFA) tienen una limitada expresión de manifestaciones extracolónicas: no muestran HCEPR y raras veces presentan tumores desmoides34.
Un problema en su manejo es que clínicamente el fenotipo de estas familias puede superponerse con el de cáncer colorrectal hereditario no polipósico (CCHNP) en familias con pocos pólipos. Otro problema en el diagnóstico de la PAFA es la falta de información en el número o localización de pólipos en las familias con pocos miembros, lo que puede hacer que se confunda con casos de cáncer colorrectal esporádico.
Generalmente, se había aceptado que la PAFA seguía un patrón autosómico dominante, al igual que la forma clásica de PAF, pero con un índice de mutaciones de novo superior. Recientemente, se ha descrito que hasta un 30% de las PAFA siguen un patrón de herencia autosómico recesivo asociado a la mutación en el gen MYH, por lo que esta alteración se denomina «poliposis asociada al MYH»39.
BASES MOLECULARES DE LA ENFERMEDAD
Se considera que más del 90% de las familias afectadas de PAF «clásica» presentan mutaciones en el gen APC en la línea germinal7. Es muy importante entender que la identificación de dicha mutación en el gen APC permite confirmar el diagnostico de PAF3.
El gen APC fue identificado hace 10 años, abarca 98 kb del ADN genómico en el cromosoma 5q21 y contiene 19 exones36. La característica más importante del gen es el gran tamaño del exón 15 (6574 pb), lo que representa el 77% de la región codificante10.
La proteína APC es un polipéptido de 2.849 aminoácidos con un peso molecular aproximado de 312 kDa, que consta de múltiples dominios, lo que permite que interaccione con un gran número de otras proteínas. Esta proteína es una parte integral de la vía de transmisión de distintas señales celulares pero también desempeña un papel en la adhesión celular, en la estabilidad del citosqueleto microtubular, en la regulación del ciclo celular y posiblemente también en fenómenos de apoptosis.
Las mutaciones germinales se localizan en toda la región codificante, si bien predominan en la mitad 5' del exón 15 y en los exones 8 y 11, y generalmente dan lugar a una proteína truncada. Las mutaciones más frecuentes son las localizadas en el codón 1309 (18%) y en el codón 1061 (12%). Recientemente, se han detectado también hasta un 10% de grandes deleciones en el gen APC en la PAF40.
Un dato muy interesante en la historia natural de esta enfermedad es que se han descrito diferentes asociaciones entre genotipo y fenotipo. Por ejemplo, los fenotipos con una muy grave expresividad clínica (presencia de más de 5.000 adenomas distribuidos por todo el colon y el recto) se asocian con mutaciones entre los codones 1250 y 1464, incluido el conocido hotspot 130926.
Además, distintas alteraciones genéticas pueden determinar las manifestaciones extracolónicas de la enfermedad. Por ejemplo, la presencia de HCEPR se asocia con la aparición de mutaciones en localizaciones específicas (entre el exón 9 y el codón 1445 del exón 15), y los tumores desmoides también (entre los codones 1444 y 1578)38.
Esta heterogeneidad se mantiene dentro y fuera de las familias, lo que sugiere que otros factores genéticos y ambientales deben influir en el curso de la enfermedad.
Recientemente, tras la demostración de la participación de otro gen, el MYH, con un patrón de herencia autosómico recesivo en el desarrollo de la PAF (generalmente en su forma atenuada), se ha refutado el dogma del patrón de herencia autosómica dominante en la PAF40,41.
El análisis genético de APC está indicado para confirmar el diagnóstico de esta enfermedad, pues permite hacer una evaluación presintomática y debe considerarse en cualquier persona con diagnóstico clínico de PAF, especialmente si tiene familiares de primer grado menores de 40 años que aún no hayan desarrollado la enfermedad3.
TRATAMIENTO
Tratamiento quirúrgico
El objetivo del tratamiento de los pacientes con PAF es evitar las consecuencias de la transformación maligna de los múltiples pólipos existentes en el colon y el recto, una vez conocida desde hace años la indefectible secuencia adenoma-carcinoma24. En el tratamiento de la PAF se debe tener en cuenta, por un lado, el potencial de malignización de los pólipos distribuidos por el área afectada y, por otro, la morbilidad asociada con las diferentes técnicas quirúrgicas.
La intervención quirúrgica ideal, sobre todo teniendo en cuenta que la mayoría de los pacientes son jóvenes y asintomáticos, sería la asociada a una baja morbilidad, con buenos resultados funcionales postoperatorios y que eliminara por completo el riesgo de carcinoma colorrectal. Desafortunadamente, no hay una técnica quirúrgica que cumpla por completo estas premisas. De esta manera, la elección de cada una de estas técnicas quirúrgicas estará en función de toda una serie de aspectos que cabe considerar: la manifestación clínica de la enfermedad, la edad del paciente, la presencia de cáncer al diagnóstico, aspectos genéticos y, finalmente, aspectos sociales, como la dificultad para el seguimiento de cada uno de los pacientes8,24.
Por otro lado, la indicación del tratamiento quirúrgico no ofrece dudas en los pacientes con diagnóstico establecido de neoplasia colorrectal, pero en los pacientes asintomáticos diagnosticados mediante marcadores moleculares en una unidad de consejo genético, el momento más adecuado de plantear la intervención es mucho más difícil de planificar. En los pacientes pertenecientes a familias con la enfermedad y un alto grado de penetrancia, se considera adecuada la intervención quirúrgica al final de la adolescencia (15-18 años)24. En otras situaciones, la cirugía profiláctica debe considerarse antes de los 25 años, o al primer diagnóstico (ante una edad más avanzada), para reducir al máximo el riesgo de cáncer colorrectal durante el seguimiento40.
Técnicas quirúrgicas
Proctocolectomía total con ileostomía terminal
La proctocolectomía total con construcción de ileostomía terminal definitiva es la técnica que menos se utiliza actualmente. Evitar la aparición de carcinoma colorrectal está asegurado, pero la morbilidad de la cirugía pélvica, así como las inevitables consecuencias psicosociales asociadas a la necesidad de la realización de un estoma, son sus principales desventajas. Actualmente, la principal indicación de dicha técnica son los casos en que el paciente presente una neoplasia de recto que por su localización cercana al margen anal no permita la restauración del tránsito intestinal24 (fig. 2). También se ha utilizado esta técnica en pacientes que precisan una colectomía total con extirpación del recto y que, por algún problema asociado (p. ej., por incontinencia fecal establecida, obesidad mórbida u otras), no permita realizar la proctocolectomía con reservorio ileal.
En los pacientes con dicha intervención, a pesar de haber eliminado la posibilidad de afección colorrectal, se deberá realizar también un seguimiento adecuado para detectar posibles manifestaciones extracolónicas42.
Colectomía total con anastomosis ileorrectal
Como esta técnica preserva el recto, su ventaja principal es la baja morbilidad postoperatoria que conlleva (p. ej., en porcentaje de disfunción sexual postoperatoria y el tipo de hábito deposicional posquirúrgico) respecto a las otras técnicas descritas. Contrariamente, su principal inconveniente es la necesidad de un seguimiento estricto para evitar las consecuencias de la aparición de nuevos pólipos o cáncer en el recto no extirpado43,44.
La identificación de los pacientes con mayor riesgo de desarrollar cáncer en el recto remanente parece ser importante, tanto para indicar una u otra técnica como para planear diferentes estrategias de seguimiento. La incidencia de cáncer de recto después de esta técnica es variable en la literatura médica clásica, que oscila entre el 10 y el 40% a los 20 años de seguimiento8,45,46. Diferentes estudios han intentado predecir la mayor o menor posibilidad de cáncer de recto durante el seguimiento. La presencia de cáncer (además de la poliposis) en la primera intervención, una anastomosis ileorrectal situada a más de 15 cm del margen anal, el número total de pólipos presentes o de nueva aparición en el recto, la presencia de pólipos planos y la edad avanzada son los factores más significativos47,48.
Los medios para reducir el riesgo de cáncer en el recto remanente en estos pacientes se basará en un adecuado calendario de seguimiento, detección y tratamiento endoscópico, de los pólipos del recto preservado24. Algunos grupos han realizado también ensayos clínicos con el objetivo de valorar el efecto del uso de distintos fármacos (p. ej., sulindaco) durante el seguimiento de estos pacientes con resultados francamente esperanzadores49,50. A pesar de ello, por la gravedad de algunos efectos secundarios del tratamiento médico e incluso porque su uso no excluye totalmente la posibilidad de transformación maligna de los pólipos en el recto preservado, no hay todavía una clara indicación de tratamiento profiláctico en estos pacientes3,24,51.
Proctocolectomía restauradora con reservorio ileal y anastomosis ileoanal
En esta técnica se realiza la resección del colon y de todo el recto, manteniendo únicamente unos 2 cm de epitelio anal transicional y mucosa distal (canal anal). De esta manera, se elimina en buena parte el riesgo de aparición de nuevos pólipos y, por tanto, el riesgo de cáncer colorrectal. Además, se acompaña de la restauración del tránsito intestinal mediante la construcción de un reservorio con íleon que actuaría funcionalmente de «neorrecto».
A lo largo de la historia se han descrito varios tipos de reservorio ileal pero, sin duda alguna, el reservorio ileal en J, descrito por Parks et al52 hace más de 25 años sigue siendo hoy en día la técnica quirúrgica más empleada53. Finalmente, la anastomosis entre el reservorio ileal y el ano es motivo de controversia, ya que puede ser mecánica o manual, en cuyo caso se realiza una exéresis de la mucosa más distal del canal anal (mucosectomía quirúrgica).
Disponer de un procedimiento quirúrgico que elimine casi todo el tumor, evitando los inconvenientes de un estoma definitivo, sólo tiene como «talón de Aquiles» la morbilidad postoperatoria asociada a esta intervención. La tasa de complicaciones postoperatorias inmediatas o durante el seguimiento y la necesidad de reintervención, aun siendo menor que en los casos de colitis ulcerosa, no es irrelevante54,55.
¿Cuál es la técnica quirúrgica más adecuada?
En la tabla III se expone un resumen de las características principales de las distintas técnicas quirúrgicas expuestas anteriormente. Parece evidente, una vez expuestas las distintas técnicas quirúrgicas, que la elección de una de ellas depende tanto de la enfermedad cómo de otros aspectos, como la edad o las expectativas del paciente hacia el tratamiento que va a recibir56-58. Esta última consideración es particularmente importante en esta enfermedad, por afectar a pacientes jóvenes, con frecuencia asintomáticos y que pueden tener experiencias más o menos desafortunadas en otros miembros familiares afectados56. La información al paciente y a la familia será especialmente importante en la toma de decisiones en esta enfermedad.

La proctocolectomía total con ileostomía definitiva es un procedimiento que se reserva únicamente para los casos de cáncer rectal bajo en los que no esté indicada la construcción de una proctocolectomía restauradora con reservorio ileal (fig. 2).
La colectomía total con anastomosis ileorrectal estare;ximo 20 pólipos) y/o con alteraciones genéticas con baja penetrancia. Siempre hay que insistir en todos los casos en el compromiso por parte del paciente de realizar los controles de seguimiento adecuados. Los pacientes con un diagnóstico tardío de la enfermedad, que tengan poca o nula afección del recto y que por una edad avanzada nos permita prever unos resultados funcionales poco satisfactorios con el reservorio ileal, también son excelentes candidatos para esta técnica quirúrgica.
La proctocolectomía total con reservorio ileal y anastomosis ileoanal debería realizarse en pacientes con formas agresivas de la enfermedad o con gran afección del recto (más de 20 pólipos adenomatosos). La realización de una mucosectomía y anastomosis manual debería indicarse sobre todo en los casos con grave afección difusa del recto hasta la línea pectínea. Se recomienda también la proctocolectomía restauradora en pacientes con síndrome de Gardner por el riesgo de aparición de tumor desmoide tras la colectomía y la anastomosis ileorrectal, que podría hacer inviable la proctocolectomía posterior. Más recientemente, se ha recomendado también esta opción terapéutica ante aparición de determinadas alteraciones genéti-cas que permitan prever una evolución tórpida de la enfermedad59.
Finalmente, es interesante apuntar que determinadas mutaciones tienen una influencia significativa en la evo lución más o menos agresiva de esta enfermedad59,60. Quizás en un futuro no muy lejano, la determinación detallada de estas alteraciones genéticas asociadas a la peor evolución de esta enfermedad (p. ej., aparición progresiva de pólipos en el recto remanente en pacientes con colectomía total e ileorrectostomía) podrá tener un peso relevante en la decisión de la técnica quirúrgica.
Papel del tratamiento médico en la poliposis adenomatosa familiar
La administración de fármacos para la quimioprevención de lesiones en estos pacientes es controvertida3. Parece que los antiinflamatorios no esteroideos (AINE), como sulindac o celecoxib, son los que han mostrado resultados más esperanzadores61-63. A pesar de ello, por un lado hay cierta evidencia que demuestra que el tratamiento con AINE en estos pacientes es inefectivo para tratar la presencia de adenomas24,64, y otros65 presentan resultados favorables tras la administración de estos fármacos. Probablemente, habrá que esperar a las conclusiones de más estudios para situar esta terapéutica en el escalón más adecuado, teniendo en cuanta además los efectos adversos comunicados con estos fármacos.
PROTOCOLO DIAGNÓSTICO Y DE SEGUIMIENTO
Las pautas de seguimiento se han diseñado tras saber que, después del tratamiento profiláctico en los pacientes con PAF, las causas más importantes de mortalidad y morbilidad son el cáncer duodenal, los tumores desmoides y, en los pacientes con colectomía y anastomosis ileorrectal, el cáncer de recto3,42,66.
Protocolo diagnóstico y seguimiento en familias con el diagnóstico genético de poliposis adenomatosa familiar
La identificación de los individuos en situación de riesgo de padecer la PAF es fundamental para disminuir la morbimortalidad de la enfermedad3,24. Desde la introducción del diagnóstico molecular predictivo se ha facilitado la identificación de los individuos en situación de riesgo. Por ello, la primera acción que se debe realizar tras el diagnóstico de PAF es remitir al paciente y su familia a una unidad de consejo genético para llevar a cabo un análisis genético y recibir consejo24,40.
Los familiares de primer grado de un paciente afectado de PAF, en riesgo de desarrollar la enfermedad, deberían iniciar un programa de cribado con una endocopia digestiva baja entre los 10 y los 15 años de edad40. Debería realizarse una rectosigmoidoscopia anual de los 12 a los 25 años de edad, cada 2 años entre los 25 y los 35 años, y cada 3 años entre los 35 y los 50 años.
Debido a que los adenomas aparecen difusamente en todo el colon, la realización de una sigmoidoscopia es suficiente para establecer si un individuo expressa la enfermedad3. Contrariamente, si se sospecha una forma atenuada (PAFA), debe realizarse una colonoscopia, por ser el colon derecho muchas veces el segmento único o más afectado.
Seguimiento en los pacientes con afección colorrectal y no operados
Se debe informar siempre de la necesidad absoluta de tratamiento quirúrgico ante el diagnóstico de la enfermedad, tal como se ha apuntado anteriormente. En cualquier caso, mientras el paciente no acepte operarse (en contra de lo indicado) hay que realizar como mínimo una colonoscopia cada 6-12 meses. Debe recomendarse también el control y el seguimiento adecuado del tracto digestivo superior84.
Seguimiento y control del recto en pacientes operados
En los pacientes portadores de una colectomía total y una anastomosis ileorrectal, debe realizarse cómo mínimo una rectoscopia cada 6-12 meses por el riesgo evidente de nuevos adenomas y/o cáncer de recto3,24. Los adenomas de pequeño tamaño deben biopsiarse en todos los casos. El incremento en el número o el tamaño de los adenomas ya presentes en el recto requiere un seguimiento más exhaustivo, y se recomienda además la exéresis de éstos si son mayores a 5 mm. La presencia de displasia grave o adenomas grandes (más de 1 cm) en el recto es una indicación absoluta para la práctica de una proctectomía24.
En los pacientes con reservorio ileoanal también deben realizarse controles periódicos. Se han descrito casos de adenomas en el manguito rectal remanente, especialmente cuando no se efectúa mucosectomía, así como aparición de adenomas en el mismo reservorio. Por todo ello, es aconsejable realizar una exploración endoscópica del reservorio en estos pacientes, como mínimo de periodicidad anual.
Seguimiento de las lesiones extracolónicas asociadas a la poliposis adenomatosa familiar
Lesiones en el tracto digestivo superior
Es importante establecer un programa de seguimiento y tratamiento de la lesiones gastroduodenales en todos los pacientes con PAF67-70. Hay un cierto consenso en los diferentes estudios respecto a que para el seguimiento debe realizarse una endoscopia digestiva alta con visión lateral, con o sin magnificación, realizando biopsias seriadas (mínimo 5) incluso ante la ausencia de pólipos a la exploración70,71. Esta última recomendación surgió a partir de la evidencia de que en algunos casos sin poliposis gastroduodenal la biopsia de la zona de la ampolla de Vater macroscópicamente normal permitió en algún caso realizar un diagnóstico de displasia71.
La primera endoscopia digestiva alta en pacientes con PAF conocida, aunque sean formas atenuadas, y/o pacientes asintomáticos, debería realizarse a la edad de 20 años o en el momento del diagnóstico de la enfermedad19,24,61. Esta primera endoscopia permitirá valorar y clasificar mediante la estadificación de Spigelman et al21 el tipo de lesiones presentes. No hay un consenso en el calendario de seguimiento ante la presencia de pólipos en el área gastroduodenal, pero un posible esquema sería el siguiente: si el paciente presenta una endoscopia normal o la presencia de una poliposis leve (estadio 0-II), la siguiente endoscopia se repetiría a los 3 años y, si no hay progresión, a los 5 años. Por otro lado, cuando aparecen grandes pólipos, lesiones numerosas o que determinen un riesgo elevado (estadio III) el seguimiento debe ser como mínimo anual (6-12 meses), valorando además la necesidad de tratamientos específicos3,70. Finalmente, hay suficiente evidencia para recomendar que ante la presencia de una displasia grave en la biopsia (estadio IV) o en determinadas lesiones periampulares grandes, se debe indicar la resección quirúrgica24,62.
Tumores desmoides
Desgraciadamente, la evolución con o sin tratamiento de los tumores desmoides es tan variable que es difícil o casi imposible realizar una pauta de seguimiento y tratamiento de estas lesiones30,42,72. Por ello, sería muy interesante describir las variables que permiten predecir su evolución, así como los métodos de clasificación.
A pesar de que el seguimiento de los tumores desmoides es controvertido, una conducta prudente sería recomendar realizar una tomografía computarizada abdominal de control anual y, por supuesto, en cualquier momento ante la sospecha diagnóstica.
Seguimiento de otras lesiones asociadas
Si la retinoscopia es normal, sólo debe repetirse cómo prueba de cribado cada 2 o 3 años en determinados ámbitos donde no hay la posibilidad de realizar un estudio genético familiar.
La ortopantomografía para el diagnóstico de un osteoma mandibular sólo debe realizarse para establecer el diagnóstico de la enfermedad familiar o si hay clínica asociada.
En general, se considera necesaria la exploración física anual (incluida la palpación cervical para diagnosticar una enfermedad tiroidea asociada) con el fin de detectar otras lesiones asociadas a la enfermedad. A pesar de ello, la utilidad de las técnicas de imagen para descartar todas las lesiones asociadas extracolónicas en la evolución de los pacientes es controvertida24,66.
PAPEL DE LAS UNIDADES DE CONSEJO GENÉTICO Y REGISTROS DE POLIPOSIS
El diagnóstico, el tratamiento y el seguimiento de los pacientes con PAF es un paradigma de colaboración multidisciplinaria entre diferentes profesionales. Para el correcto control y la aplicación de pautas asistenciales y de investigación en esta enfermedad, hay que considerar la necesidad de crear registros de pacientes con PAF. Estos registros están directamente supervisados por un equipo de distintos especialistas que discuten los casos mediante sesiones periódicas y atienden a estos pacientes o a sus familias. El objetivo fundamental es elaborar protocolos terapéuticos y diagnósticos a partir de la detección, el conocimiento y el seguimiento de las familias afectadas de la enfermedad o de riesgo mediante el consejo genético73,74. El prestigioso Hospital de St. Mark's de Londres, por ejemplo, posee desde 1925 uno de los registros más importante del mundo de pacientes con PAF, lo que ha permitido analizar una gran cantidad de información muy valiosa75.
Las unidades de consejo genético en la PAF contribuyen en el proceso de comunicación que atiende a las necesidades y preocupaciones individuales y familiares relacionadas con el desarrollo y/o la transmisión de la PAF76. De hecho, el consejo genético es el acto en el cual se informa al paciente de los riesgos que tiene de presentar una determinada enfermedad y de transmitirla, así como de los procedimientos diagnósticos disponibles tanto para él como para sus familiares3.
Específicamente, este proceso incluye la intervención de uno o más profesionales, correctamente formados, para ayudar a un individuo o familia a: a) comprender los hechos médicos, incluido el diagnóstico, la probable evolución de la enfermedad y las opciones de manejo clínico disponibles; b) comprender de qué manera la herencia contribuye en la PAF y las posibilidades de aparición de la enfermedad en otros miembros de la familia; c) entender las alternativas disponibles para el manejo del riesgo de aparición del cáncer en la PAF, es decir, medidas de prevención primaria y secundaria disponibles, incluido el test genético predictivo; d) elegir las acciones más apropiadas según el riesgo personal, las expectativas de la familia, y las convicciones éticas y religiosas propias del individuo, y actuar en consecuencia y concordancia con dicha decisión, y e) ofrecer el soporte necesario al individuo y la familia para el afrontamiento de la enfermedad. Con todo ello, el primer punto en la guía de manejo de estos pacientes comunicada por la Sociedad Americana de Cirujanos de Colon y Recto es el consejo genético, punto imperativo en la detección de nuevos casos y, por tanto, en la disminución de casos con mala evolución24.
Finalmente, ante la elevada penetrancia de la PAF y la disponibilidad de medidas preventivas eficaces, el estu dio del gen APC (test genético) puede considerarse como una «prueba ideal» y debería tenerse en cuenta como una prueba diagnóstica habitual en la práctica clínica en los individuos de riesgo73.
Se desconoce por el momento si esta estrategia de predicción de riesgo podrá decidir la mejor intervención quirúrgica o, lo que sería más deseable aún, se podrán controlar las manifestaciones clínicas de la enfermedad aplicando nuevas terapias.
CONCLUSIONES Y RECOMENDACIONES
En los últimos años los avances en genética y biología molecular han permitido conocer más profundamente la PAF. Esta enfermedad se caracteriza por una herencia autosómica dominante, aunque se ha descrito un porcentaje elevado de pacientes con mutaciones de novo u otros patrones hereditarios. Aunque la principal característica es la presencia de múltiples pólipos en el colon y el recto, hay también todo un conjunto de manifestaciones extracolónicas de la enfermedad. Sabemos que las lesiones del colon sin tratamiento evolucionarán invariablemente hacia cáncer colorrectal. La mejor técnica quirúrgica es la que permite un buen balance entre el control de la enfermedad y la morbilidad asociada a ésta. Después de realizar un adecuado consejo genético, la proctocolectomía total restauradora con reservorio ileoanal es la técnica quirúrgica de elección en las formas más graves, mientras que la colectomía total e ileorrectostomía será una alternativa valida para pacientes con formas más leves de la enfermedad. Sea cual sea el tratamiento efectuado, deberán realizarse pautas de seguimiento adecuadas tanto para el control de la enfermedad rectal como de las lesiones extracolónicas asociadas a estos pacientes. Para ello, la creación de unidades multidisciplinarias y registros de pacientes y familias afectadas de poliposis puede ser de gran ayuda. En un futuro no muy lejano los nuevos conocimientos de biología molecular contribuirán a mejorar el diagnóstico y el tratamiento, e incluso a prevenir la enfermedad.

