





Casos Clínicos en Gastroenterología y Hepatología
Más datosLa enfermedad de von Hippel-Lindau (VHL) es un síndrome familiar de baja prevalencia, con un patrón de herencia autosómico dominante. Los pacientes mayoritariamente presentan una mutación germinal en el gen supresor de tumores VHL, que predispone para la formación de tumores y quistes en diferentes órganos. La forma de presentación típica suele ser la aparición de hemangioblastomas del sistema nervioso central o la retina, carcinomas de células renales y feocromocitomas, siendo menos frecuente la afectación digestiva caracterizada por lesiones pancreáticas predominantemente quísticas. Esta puede ser la primera manifestación y preceder en años a la aparición de otras manifestaciones de la enfermedad1.
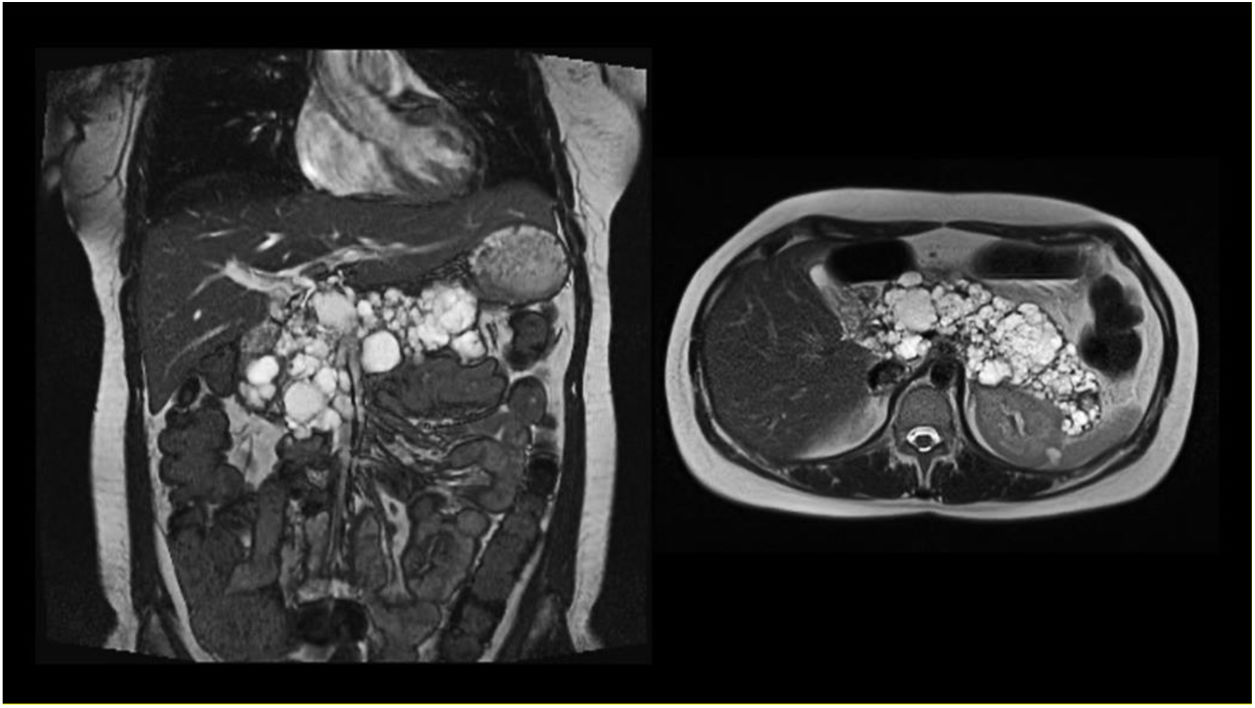
Presentamos el caso de una mujer de 30 años que se realiza una ecografía abdominal como estudio de dispepsia, identificándose múltiples quistes pancreáticos. Posteriormente se realiza TC abdominal y RMN (fig. 1) para su caracterización, visualizándose además 2 quistes renales complejos y un angiomiolipoma renal derecho. Con estos hallazgos, y dada la edad de la paciente, se sospecha radiológicamente la enfermedad de VHL, realizándose el estudio genético que confirma el diagnóstico. Además, la paciente presentaba una elastasa muy baja en heces (<50μg/g), por lo que se inició terapia enzimática sustitutiva aun en ausencia de déficits nutricionales clínicos y analíticos, con mejoría de la clínica dispéptica. El estudio familiar resultó negativo.
 que muestran múltiples quistes simples y cistoadenomas serosos pancreáticos que reemplazan prácticamente la glándula pancreática normal.")
La afectación digestiva en la enfermedad de VHL suele ser exclusivamente pancreática, y aparece en el 35-70% de los pacientes. Puede presentarse en forma de lesiones quísticas como quistes simples (70%) o cistoadenomas serosos (10-15%), y en forma de lesiones sólidas como tumores neuroendocrinos pancreáticos (pNET) (5-18%), o excepcionalmente como metástasis de un carcinoma de células renales. Algunos estudios han encontrado una asociación entre el tumor papilar mucinoso intraductal de rama lateral y la VHL1–3.
Las lesiones pancreáticas son habitualmente asintomáticas y se presentan como una afectación difusa quística del páncreas. Los casos sintomáticos (5-10%) se manifiestan: en forma de dolor abdominal o ictericia, como consecuencia de la compresión de órganos contiguos; pancreatitis aguda, como consecuencia de la secreción neuroendocrina de un pNET; clínica de insuficiencia pancreática exocrina o endocrina, por la sustitución y desaparición del parénquima pancreático4.
No se ha demostrado que los pacientes con VHL tengan un riesgo aumentado de adenocarcinoma de páncreas. Sin embargo, los pNET pueden ser una causa de mortalidad debido a su potencial maligno y riesgo de metástasis. En comparación con los pNET esporádicos, aparecen a edades más tempranas, son más frecuentemente múltiples y no funcionantes, y tienen menor riesgo de metástasis. El riesgo de metástasis se correlaciona con el tamaño tumoral, siendo excepcional en los menores de 2cm. Los pNET pueden ser sólidos o quísticos y en este último caso pueden ser difíciles de diferenciar de un quiste simple o un cistoadenoma seroso. Tanto la TC como la RMN son comparables en términos de detección de pNET. Cuando estas no son concluyentes, la realización de una ecoendoscopia con punción puede ser de utilidad en el diagnóstico diferencial. Se recomienda un seguimiento estrecho debido al riesgo de metástasis2,3.
Respecto al tratamiento de los pNET, los datos son limitados. No se recomienda el tratamiento quirúrgico de las lesiones quísticas pancreáticas salvo que sean sintomáticas. Se consideran factores de mal pronóstico la presencia de un tamaño tumoral≥3cm, la presencia de mutaciones en el exón 3 y un tiempo de duplicación tumoral≤500 días. Ante la presencia de 2 o 3 de estos factores, debe valorarse la cirugía. Sin embargo, esta debe recomendarse con precaución, teniendo en cuenta la afectación multifocal, la posible afectación por otros tumores y su impacto real sobre la supervivencia3,5.
Ante la frecuente ausencia de síntomas relacionados con las lesiones pancreáticas, las pruebas de cribado en pacientes con enfermedad de VHL siempre deben incluir la evaluación del páncreas. La enfermedad de VHL debe incluirse en el diagnóstico diferencial en pacientes con presencia de múltiples quistes pancreáticos, especialmente cuando se presentan en pacientes jóvenes y en ausencia de antecedentes de enfermedad pancreática. La sospecha diagnóstica de VHL implica realizar una historia familiar profunda y llevar a cabo un cribado genético del paciente y todos sus familiares1.

