






Las compañías farmacéuticas financian la mayoría de los estudios clínicos sobre medicamentos. Sin embargo, existen cuestiones clínicas que podrían no ser prioritarias desde el punto de vista comercial, pero que sin duda deberían ser abordadas, dada su transcendencia para los pacientes y la sociedad en general. La investigación clínica independiente representa aquí un pilar fundamental y su elemento básico son los estudios iniciados por el investigador (también conocidos por su nombre en inglés, investigator-initiated studies/trials). En estos estudios, es el investigador el que concibe la idea, desarrolla el proyecto y además actúa como promotor. La mayoría de los investigadores están familiarizados con la participación como colaboradores en los estudios promovidos por las compañías farmacéuticas; en dichos estudios, la compañía se encarga de todos los aspectos científicos, legales y, además, económicos, quedando la responsabilidad del investigador limitada fundamentalmente a la inclusión de pacientes y el cumplimiento del protocolo. Por el contrario, la puesta en marcha y el desarrollo de un estudio de investigación independiente requiere considerables recursos —de conocimientos, económicos y de tiempo— y una cuidadosa planificación por parte del investigador. En el presente manuscrito revisaremos cuáles son las principales características de los estudios iniciados por el investigador y sus diferencias fundamentales con los promovidos por la industria farmacéutica. Nos plantearemos también cuáles son sus fortalezas y sus limitaciones. Por último, propondremos algunas soluciones a los principales desafíos que plantean. Nuestro objetivo final es estimular a los potenciales investigadores a acometer el reto de llevar a cabo un proyecto de investigación clínica independiente.
Pharmaceutical companies fund most clinical trials on drugs. However, there are clinical issues that might not be a priority from a commercial point of view, but that should certainly be addressed, given their importance for patients and society in general. Independent clinical research represents a fundamental pillar here and its basic element is investigator-initiated studies/trials. In these studies, it is the researcher who conceives the idea, develops the project and also acts as the sponsor. Most researchers are familiar with participating as collaborators in studies sponsored by pharmaceutical companies. In these studies, the company is in charge of all the scientific, legal and financial aspects, leaving the responsibility of the researcher mainly limited to the inclusion of patients and compliance with the protocol. On the contrary, the start-up and development of an independent research study requires considerable resources – of knowledge, money and time – and careful planning on the part of the researcher. In this manuscript, we will review the main characteristics of the studies initiated by the researcher and their fundamental differences with those sponsored by the pharmaceutical industry. We will also outline what its strengths and limitations are. Finally, we will propose some solutions to the main challenges they pose. Our ultimate goal is to stimulate potential researchers to undertake the challenge of conducting an independent clinical research project.
La investigación es una actividad orientada a la obtención de nuevos conocimientos y su aplicación para la solución de problemas o cuestiones del día a día. La investigación científica en particular emplea los pasos del método científico para estudiar un aspecto determinado de la realidad, ya sea de manera teórica o experimental1.
Las compañías farmacéuticas financian la mayoría de los estudios clínicos sobre medicamentos, sobre todo en lo referente a los ensayos clínicos aleatorizados dirigidos a la aprobación inicial de un determinado fármaco2. Sin embargo, existe un gran número de cuestiones clínicas que podrían no ser prioritarias desde el punto de vista regulador o estrictamente comercial, pero que sin duda deberían ser abordadas, dada su transcendencia para los pacientes y la sociedad en general.
La investigación clínica independiente representa, por tanto, un pilar fundamental y su elemento básico son los estudios iniciados por el investigador, también conocidos como estudios clínicos independientes, o por su nombre en inglés: investigator-initiated studies (que abreviaremos como IIS a lo largo del presente manuscrito).
Un IIS es un estudio en el que el investigador concibe la idea, desarrolla el protocolo y actúa como promotor del estudio3 (en realidad, esta última función la suele desarrollar la entidad, habitualmente sin ánimo de lucro, a la que pertenece el investigador, como, por ejemplo, una sociedad científica o un instituto de investigación sanitaria). De este modo, las obligaciones de un investigador-promotor incluyen el espectro completo de un estudio de investigación: desde su concepción y diseño, pasando por el desarrollo y la coordinación del estudio, hasta el análisis de los datos y su publicación, y sin olvidar el aprovisionamiento y la gestión de los fondos, las responsabilidades legales del promotor ni los trámites administrativos que implican al Comité de Ética de Investigación con Medicamentos (CEIm) y a la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS).
La mayoría de los investigadores están familiarizados con la participación como colaboradores en los estudios promovidos por las compañías farmacéuticas; en dichos estudios, la compañía se encarga de todos los aspectos científicos (diseño del protocolo, etc.), legales (interacciones con las autoridades sanitarias) y, además, económicos (financiación del estudio), quedando la responsabilidad del investigador limitada en la mayoría de los casos a la inclusión de pacientes y al cumplimiento del protocolo4.
Por el contrario, la puesta en marcha y desarrollo de un IIS requiere considerables recursos —de conocimientos, económicos y de tiempo— por parte del investigador principal. Por tanto, debemos estar bien convencidos de que merece la pena embarcarnos en una misión de tal envergadura. Por ello, en el presente manuscrito revisaremos primero cuáles son las principales características de los IIS y sus diferencias fundamentales con los estudios promovidos por la industria farmacéutica. Revisaremos también cuáles son sus fortalezas y sus limitaciones. Por último, propondremos algunas soluciones a los principales desafíos que plantea la investigación clínica independiente. Nuestro objetivo final no es otro que estimular a los potenciales investigadores a acometer el reto de llevar a cabo un IIS.
¿Cuáles son las principales diferencias entre un estudio promovido por la industria y por un investigador independiente?Papel de investigadorEn la mayoría de los estudios promovidos por la industria, el investigador desarrolla una labor relativamente pasiva, limitándose sus funciones principales a reclutar a pacientes y aplicar el protocolo preestablecido. Por el contrario, en los IIS, el investigador principal desempeña un papel de liderazgo y tiene el protagonismo intelectual.
Objetivos el estudioAunque el objetivo de ambos tipos de estudios puede coincidir (de hecho, lo hace en muchos casos), la meta de los ensayos clínicos promovidos por la industria farmacéutica suele estar relacionada con el registro/aprobación de medicamentos, siendo la empresa promotora la propietaria de los datos. Por su parte, en los IIS, el objetivo es fundamentalmente científico y los resultados son propiedad de los investigadores (en realidad, de la entidad sin ánimo de lucro a la que pertenece el investigador principal y que suele actuar de promotor). Dicho de otro modo, en el primer caso se espera que el desarrollo de nuevos fármacos, además de ofrecer nuevas alternativas terapéuticas, genere beneficios económicos, mientras que en el segundo la motivación científica tiene un papel preponderante.
De este modo, los estudios promovidos por la industria buscan habitualmente la aprobación para un nuevo medicamento5,6. Es preciso señalar aquí que este objetivo de la industria contribuye muy positivamente al avance de la medicina y al mejor tratamiento de los pacientes, al ser el origen fundamental del desarrollo de nuevos fármacos. Por su parte, los IIS intentan responder preguntas relevantes de la práctica clínica que, de otra manera, es probable que no fueran cubiertas por la investigación de la industria7. Aunque ambos tipos de estudios puedan compartir el interés sobre un mismo fármaco, los IIS ayudan a generar datos sobre la efectividad y la seguridad de un medicamento ya aprobado en el entorno del mundo real (p. ej., en nuevas indicaciones o regímenes de dosificación distintos de los inicialmente aprobados). De este modo, por lo general, la atención de los IIS se centra en la optimización de los tratamientos ya existentes, incluyendo la comparación con otras estrategias terapéuticas (tanto desde el punto de vista de su efectividad como de su eficiencia) o la evaluación de la seguridad a largo plazo (recordemos que los ensayos clínicos promovidos por la industria suelen tener un seguimiento limitado en el tiempo).
En resumen, invertir en investigación clínica independiente beneficia al paciente en particular y a la sociedad en general, en términos de reducción del impacto de las enfermedades, optimización de las estrategias de atención sanitaria y contención de costes de los sistemas sanitarios. Todos estos beneficios suponen un excelente complemento de los derivados de la investigación promovida por la industria.
Criterios de inclusión en el estudioLos criterios de inclusión en los ensayos clínicos promovidos por la industria son habitualmente muy estrictos, ya que las condiciones en las que se evalúan los fármacos deben ser generalmente muy controladas y, como consecuencia, se evalúan poblaciones muy selectivas y sus resultados son difícilmente extrapolables al mundo real8. Por el contrario, los IIS deberían cubrir poblaciones más amplias y generar conocimiento sobre el papel de los tratamientos en la vida real.
Financiación del estudioEn los estudios promovidos por la industria farmacéutica, esta patrocina el ensayo y lo financia. Los costes de los ensayos clínicos suelen ser considerables, pues incluyen, entre otros: el pago a investigadores y las reuniones de estos, el diseño del proyecto de investigación, la elaboración del cuaderno de recogida de datos (CRD), la póliza de seguro, las tasas de presentación a los CEIm y a la AEMPS, el suministro del medicamento de estudio, el pago de las pruebas extraordinarias (no realizadas por práctica clínica), la preparación de informes periódicos, la monitorización, etc.9,10. Esto obliga a que, en los IIS, el investigador deba buscar financiación a través de convocatorias públicas o de las propias compañías farmacéuticas.
Beneficio económico del investigadorEn general, el interés por la investigación y el compromiso con el desarrollo científico es el principal motivo que mueve a los investigadores a participar en estudios promovidos por la industria farmacéutica. No obstante, en estos proyectos también existe un incentivo económico que compensa el esfuerzo invertido en la participación en los mismos. Sin embargo, en la mayoría de los IIS no existe este tipo de compensación económica para los investigadores (como hemos comentado anteriormente, las necesidades de estos estudios son muchas y los recursos disponibles, escasos). Por tanto, el investigador principal únicamente cuenta con el interés científico de su idea para convencer y motivar a los investigadores colaboradores de su participación en el mismo8.
Calidad de la monitorización y de los resultados del estudioLa calidad de los datos incluidos en un estudio de investigación, sea cual fuere su naturaleza, debe ser excelente, solo así se obtendrán resultados de calidad. En este sentido, se ha sugerido que, en promedio, los estudios promovidos por la industria se presentan de forma más correcta y completa a las autoridades reguladoras y llevan a cabo un seguimiento más riguroso de la documentación del estudio, así como de la monitorización de los datos, en comparación con los IIS11-13. Esto podría deberse a varios motivos: en primer lugar, la formación de los investigadores independientes en buenas prácticas clínicas es, tal vez, menor que la de los promotores de los estudios de la industria farmacéutica, más «profesionalizados»; en segundo lugar, los recursos económicos necesarios para monitorizar el desarrollo del estudio y la calidad de los datos obtenidos son muy elevados y, en general, inasumibles para los investigadores independientes.
Se ha planteado si la calidad de la evidencia generada por los estudios puede diferir en función de quién sea su promotor; en este sentido, algunos autores han comprobado que los ensayos clínicos promovidos por la industria tienen más frecuentemente un diseño doble ciego14.
Por otra parte, se ha sugerido que podría existir una mayor probabilidad de demostrar un beneficio del fármaco estudiado en los ensayos clínicos promovidos por la industria en comparación con los IIS15-19, aunque esta observación no ha podido ser confirmada por otros autores20-22. En este sentido, una reciente revisión Cochrane examinó los efectos del patrocinio de la industria en la frecuencia de resultados favorables y el riesgo de sesgo, y no encontró asociación alguna23. Finalmente, y si consideramos que la publicación de un ensayo clínico es un criterio de calidad, parece que los estudios promovidos por la industria no se materializan en publicaciones en revistas biomédicas más que los iniciados por los investigadores24. Lo que sí parece constatarse es una notificación de efectos adversos más detallada en el caso de los estudios promovidos por la industria12.
En resumen, el tipo de promotor de un estudio —industria o investigador— no debería ser per se un factor determinante de la calidad del mismo. Debemos trabajar en la cultura del método científico y los principios éticos. y tener en cuenta los recursos necesarios para la monitorización del desarrollo de los estudios y de los datos registrados para que la calidad de los IIS sea tan buena como la de los estudios promovidos por la industria. Es innegable que la transparencia y la publicación abierta de los datos originales en ambos tipos de estudios incrementará su calidad y los hará más comparables.
¿Cuáles son las ventajas fundamentales de la investigación clínica independiente?Independencia de la industria farmacéuticaNo cabe duda de que una de las ventajas de los IIS es que nos ofrece la posibilidad de ser independientes de la industria a lo largo de todo el proceso del estudio, incluyendo el diseño, el análisis y la interpretación de los resultados, el establecimiento de las conclusiones y, por último, la publicación en la correspondiente revista biomédica25; y todo esto independientemente de que creamos, a priori, que los resultados vayan a ser positivos o negativos para la industria farmacéutica e independientemente también de que finalmente los resultados del estudio beneficien o perjudiquen al propietario del fármaco en cuestión.
Protagonismo de los investigadores en el estudioLos IIS ofrecen a los investigadores principales de los mismos la oportunidad de ser los auténticos líderes de un proyecto de investigación, con el consiguiente estímulo personal e intelectual que ello conlleva. Además, es probable que el impacto curricular de la publicación de un artículo de investigación clínica independiente sea superior al de otro promovido por la industria, al interpretarse como más meritorio o con mayor protagonismo científico26.
Relevancia clínica de la investigaciónSin duda, los resultados de un ensayo clínico promovido por la industria pueden tener una enorme relevancia clínica para los pacientes, que se beneficiarán del desarrollo de nuevos fármacos que sean eficaces. Sin embargo, existen aspectos muy relevantes para los pacientes y, por ende, para el investigador, que nunca serían abordados si no fuera por los IIS. Explorar aspectos clínicos que quizá no interesan —comercialmente hablando— a los estudios promovidos por la industria permite completar de forma adecuada el espectro de investigación de un determinado fármaco27.
Extrapolación de los resultados a la práctica clínicaComo se ha mencionado previamente, debido a la habitual amplitud y laxitud de los criterios de inclusión de los IIS (en contraposición al carácter más restrictivo de los estudios de la industria), los resultados que provienen de la investigación independiente se pueden extrapolar razonablemente a la práctica clínica.
Creación de sinergias con otros investigadoresLa organización de un IIS, que habitualmente requiere un diseño multicéntrico, implica el establecimiento de una infraestructura que, tras la finalización del ensayo en cuestión, puede seguir siendo de gran utilidad para la realización de futuros estudios28,29. La continua presión selectiva sobre los investigadores (en especial en España) ha promovido una evolución simbiótica que nos ha permitido sobrevivir a dicha presión ambiental30. Uno de los ejemplos más representativos de esta colaboración es la de los Centros de Investigación Biomédica en Red (CIBER) (y en concreto el de Enfermedades Hepáticas y Digestivas [CIBEREHD]), del Instituto de Salud Carlos III (ISCIII). De hecho, la creciente colaboración en red es una de las más importantes fortalezas de la gastroenterología española30. Si se puede extraer una conclusión de la experiencia española, es que la colaboración entre instituciones y redes de investigadores es la estrategia más rentable para mejorar la cantidad y, lo que es más importante, la calidad de la investigación. Es indudable que trabajar en equipo produce grandes resultados. La unión hace la fuerza y, en investigación, esto es especialmente así1,31.
Cuando nos plateemos realizar un IIS multicéntrico, deberíamos considerar que en muchas ocasiones cuesta casi el mismo esfuerzo organizarlo con unos pocos centros de nuestro entorno (p. ej., nuestra ciudad) que llevarlo a cabo a nivel nacional (contando con múltiples comunidades autónomas). Obviamente esto no siempre es así, pero con frecuencia compensa el esfuerzo extra de ampliar fronteras por los beneficios de una mayor celeridad en obtener resultados y, sobre todo, el incremento en la relevancia y el impacto científico de los mismos. A su vez, la organización de un IIS puede fortalecer el equipo investigador del centro promotor, facilitar su financiación y, en definitiva, contribuir a su sostenibilidad.
¿Cuáles son los retos fundamentales de la investigación independiente y sus posibles soluciones?Los IIS se enfrentan a una serie de barreras para poder llevarse a cabo, que en general son similares en cualquier lugar del mundo5,32,33. A continuación, se revisan dichas limitaciones y se proponen cuáles podrían ser sus posibles soluciones.
Limitación de tiempo para investigarConcebir, organizar y llevar a cabo un IIS supone un ingente esfuerzo y requiere una dedicación de tiempo muy importante, tanto para las tareas burocráticas como para las de coordinación científica34. En la tabla 1 se resumen los numerosos trámites legales y administrativos necesarios para poner en marcha un ensayo clínico35.
Trámites legales y administrativos para poner en marcha un ensayo clínico
| Escritura del protocolo de investigación |
| Diseño del cuaderno de recogida de datos |
| Redacción del consentimiento informado |
| Tramitación de la aprobación del CEIm |
| Solicitud de la aprobación de la AEMPS |
| Solicitud del número EudraCT |
| Contratación del seguro de responsabilidad |
| Preparación del archivo del promotor |
| Preparación del archivo del investigador |
| Identificación de los centros participantes |
| Negociación y firma de contratos con los centros |
| Gestión de la medicación de estudio |
| Selección del equipo de monitorización |
| Manejo de datos y estudio estadístico |
| Inclusión de pacientes en el estudio |
| Comunicación del cierre de los centros al CEIm |
| Comunicación del fin del estudio a la AEMPS |
| Elaboración del informe final |
| Publicación de los resultados en revistas biomédicas |
AEMPS: Agencia Española de Medicamentos y Productos Sanitarios; CEIm: Comité de Ética de Investigación con medicamentos.
Para los médicos que trabajan en los hospitales y que, por tanto, desarrollan una intensa labor asistencial, la planificación de un ensayo clínico independiente puede parecer casi impensable. Si el hospital es universitario y el profesional tiene funciones docentes añadidas, se convierte en misión —casi— imposible. Cuando hablamos de la necesidad de tiempo para el investigador nos referimos no solo al desarrollo del estudio en sí, sino también a los pasos previos (diseño del estudio, obtención de financiación y aprobaciones) y posteriores (contacto con los investigadores, coordinación del estudio, evaluación de resultados y publicación de los mismos). De este modo, los IIS representan para el médico-investigador un aumento muy considerable de carga laboral sin remuneración asociada.
La disponibilidad y la liberación de tiempo para investigar de los clínicos es sin duda una asignatura pendiente. Y lo seguirá siendo hasta que no logremos cambiar el paradigma actual vigente en nuestras instituciones, en las que se considera la investigación como un mero apéndice menor de la actividad asistencial (y, por tanto, prescindible y accesorio) para reconocerlo como un integrante esencial de la misma, también a nivel organizativo. Esto se logra de 2maneras: por una parte, liberando de carga asistencial al clínico y, por otra, delegando tareas de la gestión del proyecto de investigación en personal técnico cualificado que a buen seguro las llevarán a cabo mejor que él mismo.
Desde el ISCIII, gestor de la investigación biomédica en nuestro país, se reconoce esta necesidad y se intenta paliar mediante «Contratos de intensificación de la actividad investigadora»; sin embargo, las posibilidades de éxito en estas convocatorias son escasas por la dura competencia y no solucionan el problema de la mayoría de los investigadores clínicos asistenciales. La liberación de parte de la jornada mediante fondos propios puede ser una solución para algunos investigadores: el dinero nos permite comprar tiempo1. Y esto nos lleva precisamente a la que probablemente es la limitación más relevante para desarrollar investigación independiente: la dificultad para obtener financiación.
Dificultades para obtener financiaciónLa investigación clínica de alta calidad es costosa, entre otras cosas porque generalmente requiere un diseño multicéntrico y la inclusión de un número considerable de pacientes. Este elevado coste constituye la razón principal por la cual la industria farmacéutica financia la mayoría de los ensayos clínicos, quedando habitualmente los diseños menos ambiciosos como única opción al alcance de los investigadores independientes.
Las convocatorias públicas son fundamentales para obtener financiación para poder llevar a cabo investigación independiente36,37. En España, por ejemplo, este tipo de investigación está financiada por convocatorias competitivas públicas promovidas por instituciones como el ISCIII. En concreto, desde hace años existe una convocatoria específica para proyectos de investigación clínica independiente («Subvenciones para el fomento de la investigación clínica independiente, con medicamentos de uso humano o terapias avanzadas, mediante la financiación de proyectos no promovidos por la industria farmacéutica»). Tal y como se expresa en dicha convocatoria, son susceptibles de financiación por ella los proyectos dirigidos al desarrollo de ensayos clínicos (preferentemente en fase i, ii, iii) que permitan objetivar avances tangibles para los pacientes y que proporcionen evidencias a las autoridades sanitarias para su implantación en el Sistema Nacional de Salud. Y pueden ser beneficiarios de esta actuación tanto los institutos de investigación sanitaria (a los que se les presta una especial consideración), como otras entidades e instituciones sanitarias públicas o privadas (en este caso, sin ánimo de lucro y vinculadas o concertadas con el Sistema Nacional de Salud) y el CIBER. En los proyectos presentados, la figura del promotor debe recaer de forma exclusiva en la entidad gestora de la institución solicitante. El presupuesto destinado a esta convocatoria en el presente año ha sido de aproximadamente 19 millones de euros y el plazo de ejecución de los proyectos para dicha financiación es de 4años.
Las convocatorias públicas son altamente competitivas, por lo que es difícil obtener financiación en ellas. Por ello es preciso explorar otras alternativas, entre las que destaca la obtención de fondos de la propia industria farmacéutica, a través de la convocatoria de estudios iniciados por investigador. Una encuesta realizada recientemente a 28 compañías farmacéuticas y biotecnológicas reveló que el 90% de ellas disponía de este tipo de convocatorias de investigación independiente32. En este tipo de estudios, el investigador es el promotor (y, por tanto, asume las responsabilidades legales) y el laboratorio únicamente brinda apoyo financiero (si bien, en ocasiones, se limita a proporcionar el fármaco en estudio). Este tipo de subvención se utiliza como apoyo a la realización de estudios preclínicos y clínicos (incluidos los intervencionistas y no intervencionistas), que involucren de alguna manera al producto activo de la compañía farmacéutica en cuestión. El estudio debería ser realmente independiente, es decir, que la concepción del mismo haya partido espontáneamente del investigador y no haber existido «inducción» alguna por parte de la compañía farmacéutica a investigar algún aspecto sobre el que esta tenga un interés especial y de ese modo pretenda utilizar al investigador «independiente» como un mero implementador de su idea. Por tanto, al menos idealmente, si la empresa se beneficiará o no de los resultados del estudio no debería ser un criterio de concesión por parte de esta38,39. Obviamente, esta filosofía de colaboración empresa-investigador es perfectamente compatible con que la compañía farmacéutica se vea beneficiada, por ejemplo, al obtener información sobre el empleo de su fármaco en otras enfermedades no aprobadas previamente, o con esquemas posológicos no incluidos en ficha técnica, o simplemente en pacientes tratados en práctica clínica rutinaria.
Por último, es preciso mencionar que la investigación financiada y promovida por la industria farmacéutica y la investigación independiente son complementarias. En este sentido, nuestra filosofía —la de todo nuestro grupo— se basa en que la totalidad de los fondos obtenidos de proyectos deben reinvertirse en investigación, ya sea en recursos humanos o logísticos, lo que hace posible que el grupo sea sostenible a largo plazo.
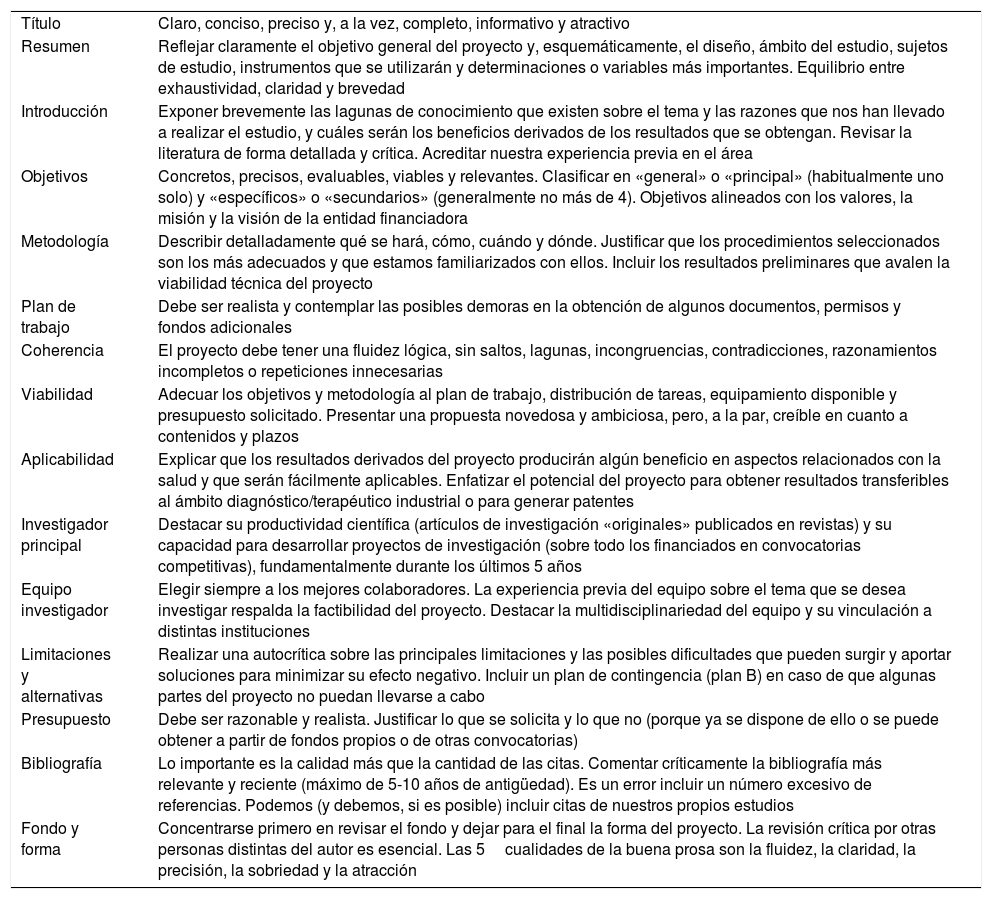
La elaboración del proyecto de investigaciónEl primer obstáculo con el que el investigador se enfrenta al desarrollar un IIS es la correcta elaboración del proyecto de investigación40. Ser capaz de preparar un buen proyecto de investigación se convierte en una exigencia para poder competir, con garantías de éxito, para obtener financiación. El proyecto de investigación es un plan escrito minuciosamente que contempla todos los aspectos, científicos, éticos y logísticos del estudio que nos proponemos llevar a cabo. Dicho proyecto tiene como objetivo presentar y describir detalladamente lo que se va a investigar, la base teórica conceptual, los componentes metodológicos y los recursos humanos, técnicos y económicos necesarios para realizar la investigación. El proyecto de investigación constituye la culminación de todo el trabajo realizado en la etapa de planificación de la investigación, por lo que representa una guía para los investigadores durante el desarrollo del estudio. Redactar un buen proyecto de investigación es la mejor garantía de que este llegará a buen puerto. En la tabla 2 se incluye una serie de recomendaciones para elaborar y presentar adecuadamente un proyecto de investigación41.
Puntos clave para elaborar adecuadamente un proyecto de investigación
| Título | Claro, conciso, preciso y, a la vez, completo, informativo y atractivo |
| Resumen | Reflejar claramente el objetivo general del proyecto y, esquemáticamente, el diseño, ámbito del estudio, sujetos de estudio, instrumentos que se utilizarán y determinaciones o variables más importantes. Equilibrio entre exhaustividad, claridad y brevedad |
| Introducción | Exponer brevemente las lagunas de conocimiento que existen sobre el tema y las razones que nos han llevado a realizar el estudio, y cuáles serán los beneficios derivados de los resultados que se obtengan. Revisar la literatura de forma detallada y crítica. Acreditar nuestra experiencia previa en el área |
| Objetivos | Concretos, precisos, evaluables, viables y relevantes. Clasificar en «general» o «principal» (habitualmente uno solo) y «específicos» o «secundarios» (generalmente no más de 4). Objetivos alineados con los valores, la misión y la visión de la entidad financiadora |
| Metodología | Describir detalladamente qué se hará, cómo, cuándo y dónde. Justificar que los procedimientos seleccionados son los más adecuados y que estamos familiarizados con ellos. Incluir los resultados preliminares que avalen la viabilidad técnica del proyecto |
| Plan de trabajo | Debe ser realista y contemplar las posibles demoras en la obtención de algunos documentos, permisos y fondos adicionales |
| Coherencia | El proyecto debe tener una fluidez lógica, sin saltos, lagunas, incongruencias, contradicciones, razonamientos incompletos o repeticiones innecesarias |
| Viabilidad | Adecuar los objetivos y metodología al plan de trabajo, distribución de tareas, equipamiento disponible y presupuesto solicitado. Presentar una propuesta novedosa y ambiciosa, pero, a la par, creíble en cuanto a contenidos y plazos |
| Aplicabilidad | Explicar que los resultados derivados del proyecto producirán algún beneficio en aspectos relacionados con la salud y que serán fácilmente aplicables. Enfatizar el potencial del proyecto para obtener resultados transferibles al ámbito diagnóstico/terapéutico industrial o para generar patentes |
| Investigador principal | Destacar su productividad científica (artículos de investigación «originales» publicados en revistas) y su capacidad para desarrollar proyectos de investigación (sobre todo los financiados en convocatorias competitivas), fundamentalmente durante los últimos 5 años |
| Equipo investigador | Elegir siempre a los mejores colaboradores. La experiencia previa del equipo sobre el tema que se desea investigar respalda la factibilidad del proyecto. Destacar la multidisciplinariedad del equipo y su vinculación a distintas instituciones |
| Limitaciones y alternativas | Realizar una autocrítica sobre las principales limitaciones y las posibles dificultades que pueden surgir y aportar soluciones para minimizar su efecto negativo. Incluir un plan de contingencia (plan B) en caso de que algunas partes del proyecto no puedan llevarse a cabo |
| Presupuesto | Debe ser razonable y realista. Justificar lo que se solicita y lo que no (porque ya se dispone de ello o se puede obtener a partir de fondos propios o de otras convocatorias) |
| Bibliografía | Lo importante es la calidad más que la cantidad de las citas. Comentar críticamente la bibliografía más relevante y reciente (máximo de 5-10 años de antigüedad). Es un error incluir un número excesivo de referencias. Podemos (y debemos, si es posible) incluir citas de nuestros propios estudios |
| Fondo y forma | Concentrarse primero en revisar el fondo y dejar para el final la forma del proyecto. La revisión crítica por otras personas distintas del autor es esencial. Las 5cualidades de la buena prosa son la fluidez, la claridad, la precisión, la sobriedad y la atracción |
Otro reto al que se enfrenta el investigador en las fases iniciales de un IIS es el diseño del CRD35. Aunque pueda no parecerlo, esta es una fase crucial de cuya calidad dependerá en buena medida la calidad de los resultados finalmente obtenidos. Dicho de otro modo, un CRD mal diseñado recogerá, sin duda, datos de mala calidad. El CRD debe estar basado en el protocolo (de este modo, si falta información en el CRD es que el protocolo está incompleto). Se debe hacer el máximo esfuerzo en seleccionar todos los datos que sean relevantes para el estudio, pero solo los imprescindibles. Por drástica que parezca, esta debe ser la regla de oro del diseñador del CRD y debe estar preparado a mantenerla, porque recibirá constantes invitaciones a violarla. Cantidad y calidad son aquí, con frecuencia, incompatibles. Se deben evitar preguntas redundantes o duplicidades. No debemos solicitar datos que pueden extraerse de otros ya incluidos. Se debe emplear un lenguaje sencillo y claro, con frases cortas y sin ambigüedades, evitando en lo posible las abreviaturas. Es importante que, antes de darlo por definitivo, el CRD sea revisado por diversos miembros del equipo investigador (además de, obviamente, el investigador principal): monitores, gestores, estadísticos, etc. Efectuar cambios significativos en el CRD cuando el ensayo clínico está ya en marcha es costoso y arriesgado, por lo que merece la pena invertir tiempo en hacerlo bien desde el principio. Finalmente, cabe señalar que un buen CRD facilitará notablemente las labores de monitorización ulteriores42.
El advenimiento de los CRD electrónicos ha supuesto un importante avance en este campo. Llegados a este punto, nos parece relevante hacer un breve comentario sobre REDCap (https://www.project-redcap.org/), acrónimo de Research Electronic Database Capture43. En particular, la plataforma on-line de investigación colaborativa Asociación Española de Gastroenterología-REDCap es un software informático que permite el diseño, el desarrollo y la gestión de CRD electrónicos en estudios de investigación. Se trata de una herramienta de uso intuitivo (tanto para los investigadores como para los monitores), ofreciendo utilidades y servicios superiores a los de la mayoría de contratistas privados. Además, provee de las herramientas necesarias para el análisis eficiente de datos, generación de estadísticas on-line, monitorización, recogida de datos en dispositivos móviles y multitud de funciones extra. Esta plataforma reduce significativamente los costes de un CRD tradicional y favorece, por tanto, el desarrollo de proyectos promovidos por los investigadores, independientemente de los intereses de la industria privada (de hecho, el uso del software es restringido, estando permitido únicamente para investigación no comercial).
Identificación de centros participantes y selección de colaboradoresDebemos elegir siempre a los mejores colaboradores, convenciéndolos de que nuestro liderazgo es una garantía de éxito para los proyectos que se desarrollen conjuntamente. Debemos seleccionar cuidadosamente el número de colaboradores; hay quien afirma que «el éxito de un proyecto de investigación generalmente está inversamente relacionado con el número de personas de quien depende». Lo que es seguro es que, como casi siempre, la calidad, y no la cantidad, es lo que importa. Se debe comprobar que el investigador colaborador está cualificado por formación y experiencia, y dispone de los recursos adecuados para realizar el ensayo clínico, así como la idoneidad de las instalaciones del centro donde se pretende llevar a cabo35. Lo más importante es seleccionar a investigadores comprometidos, con garantías de que, por ejemplo, reclutarán el número de pacientes inicialmente pactado. Es evidente que este compromiso no siempre es fácil de cumplir, pues depende de diversos factores, algunos de ellos fuera de nuestro control44. Sin embargo, es importante elegir colaboradores que midan bien sus capacidades ya que, si finalmente no pueden cumplir con lo acordado, pueden afectar seriamente la viabilidad del proyecto1.
Aprobación y clasificación por la Agencia Española de Medicamentos y Productos SanitariosLa AEMPS tiene como misión garantizar a la sociedad, desde la perspectiva de servicio público, la calidad, seguridad, eficacia y correcta información de los medicamentos y productos sanitarios en el más amplio sentido, desde su investigación hasta su utilización, en interés de la protección y promoción de la salud. La función fundamental de la AEMPS es ofrecer garantías sobre medicamentos y productos sanitarios, promoviendo el conocimiento científico-técnico. Es la autoridad sanitaria de referencia en materia de garantías de calidad, seguridad, eficacia e información de los medicamentos.
Con respecto al proceso de clasificación de los estudios de investigación, de forma muy resumida mencionaremos que, si el estudio tiene un diseño experimental, se clasificará como ensayo clínico (véase el Real Decreto 1090/2015, de 4 de diciembre, por el que se regulan los ensayos clínicos con medicamentos, los Comités de Ética de la Investigación con medicamentos y el Registro Español de Estudios Clínicos). Recordemos que un ensayo clínico es todo aquel estudio que cumple cualquiera de las siguientes condiciones: 1) asignación del sujeto a una estrategia terapéutica determinada, que no forma parte de la práctica clínica habitual; 2) la decisión de prescribir los medicamentos en investigación se toma junto con la de incluir al sujeto en el estudio clínico, y 3) se aplican procedimientos de diagnóstico o seguimiento a los sujetos de ensayo que van más allá de la práctica clínica habitual.
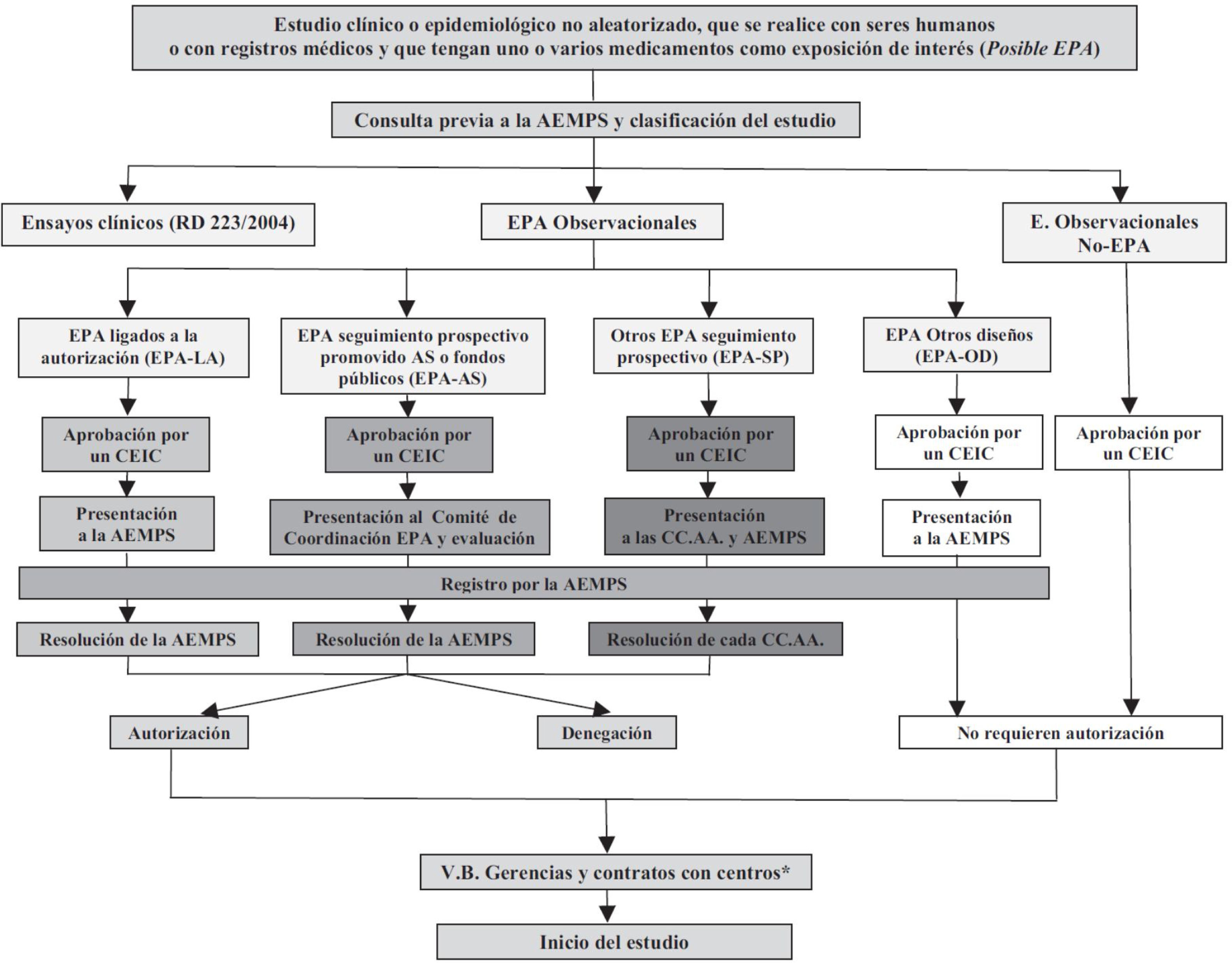
Por otra parte, de acuerdo con la Orden SAS/3470/2009, de 16 de diciembre, por la que se publican las directrices sobre estudios postautorización (EPA) de tipo observacional para medicamentos de uso humano, la AEMPS realiza una clasificación de todos los posibles EPA con medicamentos de uso humano. Dentro de estos se incluyen todos aquellos estudios clínicos o epidemiológicos no aleatorizados, realizados con seres humanos o con registros médicos, y que recojan información sobre medicamentos. Merece la pena revisar muy brevemente las principales características y requerimientos de las autoridades sanitarias para los 4tipos de EPA, cuyas vías administrativas quedan resumidas en la figura 1.
. Referencia: BOE Núm. 310, viernes 25 de diciembre del 2009, Sec. I, p. 109775: Orden SAS/3470/2009, de 16 de diciembre, por la que se publican las directrices sobre estudios postautorización de tipo observacional para medicamentos de uso humano. APA-AS: estudio postautorización promovido por las Administraciones Sanitarias (AS) o financiado con fondos públicos; EPA-LA: estudios postautorización cuya realización tiene lugar a instancia de las autoridades reguladoras y ligada a la autorización de comercialización (Agencia Española de Medicamentos y Productos Sanitarios o Agencia Europea de Medicamentos); se incluyen en esta vía administrativa tanto los estudios ligados a la autorización como los estudios de seguridad a requerimientos de las autoridades sanitarias y a los incluidos en los planes de gestión de riesgos; EPA-OD: estudios postautorización con diseño diferente al de seguimiento prospectivo, por ejemplo, estudios transversales o retrospectivos; EPA-SP: estudio postautorización de seguimiento prospectivo que no corresponde a ninguna de las categorías anteriores; No-EPA: estudios en los que el factor de exposición fundamental investigado no es un medicamento; por ejemplo, estudios de incidencia o de prevalencia de enfermedades, etc.")
Rutas administrativas de los estudios postautorización (EPA).
Referencia: BOE Núm. 310, viernes 25 de diciembre del 2009, Sec. I, p. 109775: Orden SAS/3470/2009, de 16 de diciembre, por la que se publican las directrices sobre estudios postautorización de tipo observacional para medicamentos de uso humano.
APA-AS: estudio postautorización promovido por las Administraciones Sanitarias (AS) o financiado con fondos públicos; EPA-LA: estudios postautorización cuya realización tiene lugar a instancia de las autoridades reguladoras y ligada a la autorización de comercialización (Agencia Española de Medicamentos y Productos Sanitarios o Agencia Europea de Medicamentos); se incluyen en esta vía administrativa tanto los estudios ligados a la autorización como los estudios de seguridad a requerimientos de las autoridades sanitarias y a los incluidos en los planes de gestión de riesgos; EPA-OD: estudios postautorización con diseño diferente al de seguimiento prospectivo, por ejemplo, estudios transversales o retrospectivos; EPA-SP: estudio postautorización de seguimiento prospectivo que no corresponde a ninguna de las categorías anteriores; No-EPA: estudios en los que el factor de exposición fundamental investigado no es un medicamento; por ejemplo, estudios de incidencia o de prevalencia de enfermedades, etc.
«EPA ligados a la autorización» (EPA-LA): se trata de EPA cuya realización tiene lugar a instancia de las autoridades reguladoras y ligada a la autorización de comercialización (por parte de la AEMPS o la Agencia Europea de Medicamentos). Se incluyen en esta vía administrativa los estudios de seguridad a requerimiento de las autoridades sanitarias y los estudios incluidos en los planes de gestión de riesgos de las compañías farmacéuticas.
«EPA financiado con fondos públicos o promovido por autoridades sanitarias» (EPA-AS): representa la vía administrativa típica de un ensayo clínico financiado con fondos del ISCIII, o cuando la propia AEMPS es promotora del estudio. Estos estudios requieren autorización, y no solo clasificación, por parte de la AEMPS. Esta vía tiene la ventaja de requerir un procedimiento simplificado para su autorización y de contar con una única resolución, emitida por la AEMPS en un plazo inferior a 30 días.
«EPA de seguimiento prospectivo» (EPA-SP): se refiere a estudios en los que los pacientes se seleccionan por su exposición y se siguen durante el tiempo suficiente, siendo el periodo de estudio posterior al inicio de la investigación. Son susceptibles de ser utilizados como instrumento para la inducción a la prescripción y por ello la normativa es aquí especialmente rigurosa: el estudio debe ser autorizado por todas las comunidades autónomas donde se vaya a realizar, cuyos órganos competentes resolverán en un plazo máximo de 90 días. Esta vía administrativa incluye el pago de tasas a cada una de las comunidades autónomas, lo que puede incrementar notablemente el presupuesto del estudio.
En comparación con los investigadores independientes, habitualmente la industria tiene una mayor disponibilidad de recursos humanos para llevar a cabo los farragosos trámites que requieren la aprobación y posterior gestión de los proyectos de investigación, sobre todo si se trata de un ensayo clínico multicéntrico45,46. La gestión de un estudio clínico es más complicada de lo que cabría pensar, ya que intervienen muchos actores (promotores, investigadores, hospitales, comités éticos, autoridades sanitarias, departamentos legales, monitores, pacientes, etc.). Además, es preciso trabajar según las normas de buenas prácticas clínicas, una serie de requisitos que deben cumplirse y que garantizan la protección de los derechos, la seguridad y el bienestar de los sujetos del ensayo, así como la fiabilidad de los resultados del mismo.
La gestión de los estudios se suele confiar a una Organización de Investigación por Contracto o Contract Research Organization (CRO, por sus siglas en inglés). Dicha CRO actúa de puente entre el promotor y el resto de los actores que intervienen en la realización del estudio. Los servicios que puede ofrecer una CRO se pueden dividir según la fase en la que se encuentre el estudio. En la puesta en marcha incluye el desarrollo y la revisión del protocolo de investigación, la adaptación de la documentación a la legislación española, la obtención de las aprobaciones necesarias por parte de los comités éticos de investigación y de las autoridades sanitarias, el diseño del CRD, la selección de los investigadores y la negociación de los contratos con los centros. Una vez que se cuenta con la aprobación y comienza el ensayo clínico, la CRO ofrece sus servicios para llevar a cabo la monitorización y farmacovigilancia. Finalmente, la CRO se encarga de la gestión de los datos obtenidos del estudio, la generación de informes y el control y almacén de la documentación.
Spanish Clinical Research Network (SCReN) es una plataforma de apoyo a ensayos clínicos multicéntricos, sin ánimo de lucro, que agrupa a 31 Unidades de Investigación Clínica y Ensayos Clínicos de 13 comunidades autónomas. SCReN colabora con investigadores, grupos cooperativos, institutos de investigación, etc., en la realización de estudios (fundamentalmente ensayos clínicos con medicamentos) independientes y multicéntricos, facilitando la gestión y la coordinación de estudios, priorizando aquellos con financiación concedida en el programa de Acción Estratégica en Salud. SCReN está financiada por el ISCIII y por los Fondos Europeos de Desarrollo Regional (FEDER).
Algunas sociedades científicas, como es el caso del Grupo Español de Trabajo en Enfermedad de Crohn y Colitis Ulcerosa (GETECCU), ponen a disposición de sus socios una secretaría científica que realiza funciones de coordinación/gestión de ensayos clínicos, en este caso sobre enfermedad inflamatoria intestinal. Dicha figura tiene como misión la gestión de todos los aspectos organizativos relacionados con los estudios de investigación multicéntricos de GETECCU, principalmente ensayos clínicos con medicamentos.
Finalmente, una opción muy atractiva, aunque de una cierta complejidad organizativa, es disponer en el propio grupo de investigación de personal entrenado para llevar a cabo todas las funciones de gestión, diseño de CRD y monitorización previamente mencionadas para los estudios de investigación independiente. La ventaja fundamental de esta alternativa es que dicho personal estará familiarizado con la enfermedad estudiada, los procedimientos habituales y la idiosincrasia del grupo de investigación, y por ello su coordinación con este debería ser excelente. Esta opción es especialmente productiva y rentable cuando el grupo tiene una intensa actividad investigadora y de una cierta continuidad. En nuestra experiencia, el entrenamiento del personal en proyectos previos del grupo repercute positivamente en los futuros, incrementando progresivamente su eficiencia. Obviamente, esto supone inicialmente una inversión de futuro y exige ser capaz de asumir la contratación de este personal especializado y de mantenerlo a lo largo del tiempo.
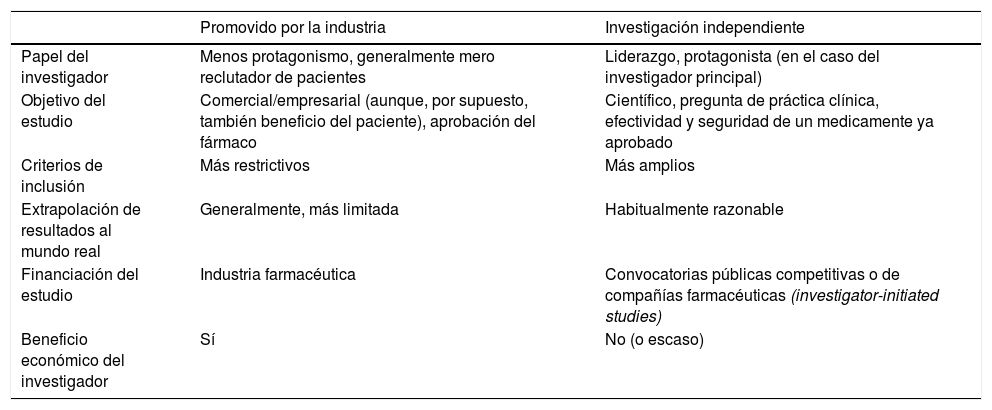
Reflexiones finalesLa investigación representa un pilar fundamental de la actividad médica y es evidente que la mayor calidad asistencial surge de la integración de una práctica clínica y una actividad investigadora excelentes. Ser investigador supone, muy especialmente en nuestro medio, un auténtico reto. La investigación independiente desempeña un papel vital en la generación de evidencia científica y complementa a los ensayos clínicos promovidos por la industria farmacéutica; aborda preguntas clínicamente relevantes para la atención del paciente, preguntas que generalmente no son el foco principal de la investigación con interés comercial. De ahí que sea fundamental desarrollar esta investigación clínica independiente que aborde temas de interés médico, aunque no genere beneficios económicos empresariales. En el presente manuscrito hemos revisado las principales características de la investigación clínica independiente, mencionando algunas diferencias entre un estudio promovido por la industria y por un investigador independiente, como se resume en la tabla 3.
Principales diferencias entre un estudio promovido por la industria y por un investigador independiente
| Promovido por la industria | Investigación independiente | |
|---|---|---|
| Papel del investigador | Menos protagonismo, generalmente mero reclutador de pacientes | Liderazgo, protagonista (en el caso del investigador principal) |
| Objetivo del estudio | Comercial/empresarial (aunque, por supuesto, también beneficio del paciente), aprobación del fármaco | Científico, pregunta de práctica clínica, efectividad y seguridad de un medicamente ya aprobado |
| Criterios de inclusión | Más restrictivos | Más amplios |
| Extrapolación de resultados al mundo real | Generalmente, más limitada | Habitualmente razonable |
| Financiación del estudio | Industria farmacéutica | Convocatorias públicas competitivas o de compañías farmacéuticas (investigator-initiated studies) |
| Beneficio económico del investigador | Sí | No (o escaso) |
Es evidente que existe un déficit de investigadores con experiencia en la realización de estudios independientes. Muchos investigadores dominan ciertos conceptos sobre metodología de la investigación clínica, pero la organización y el desarrollo de un ensayo clínico exige el conocimiento de muchos otros aspectos: elaboración de protocolos, diseño de CRD, consideraciones éticas y legales, solicitudes a las autoridades sanitarias y comités de ética, monitorización de la calidad de los datos, farmacovigilancia y un largo etcétera. Si a esta notable complejidad organizativa y burocrática le sumamos la dificultad para obtener financiación y el nulo incentivo económico (en contraste con los estudios promovidos por la industria), no es de extrañar que el número de IIS sea más bien escaso. Sin embargo, la investigación independiente se asocia también con grandes ventajas, como se resume en la tabla 4, entre los que destaca el papel protagonista y de liderazgo del investigador y la relevancia clínica de los resultados obtenidos.
Ventajas de la investigación clínica independiente
| Oportunidad de independencia de la industria farmacéutica |
| Mayor imparcialidad, menos conflictos de interés |
| Papel protagonista y liderazgo del investigador principal y del equipo investigador |
| Relevancia clínica de los resultados en escenarios no cubiertos por la investigación de la industria farmacéutica |
| Extrapolación razonable de resultados a la práctica clínica |
| Creación de sinergias con otros investigadores |
| Fortalecimiento del propio equipo investigador |
Es indudable que la investigación clínica independiente debe enfrentarse a numerosos retos que hemos revisado en el presente manuscrito, pero también hemos sugerido cuáles podrían ser, al menos en algunos casos, sus posibles soluciones. Es evidente también que se requiere el compromiso gubernamental y de las autoridades sanitarias para facilitar e incentivar el desarrollo de IIS, estimulando y formando investigadores independientes, incrementando la financiación de las convocatorias públicas competitivas y desarrollando infraestructuras de apoyo a dicha investigación.
La actividad investigadora independiente cierra el círculo que se inicia con la detección de una necesidad clínica no cubierta en nuestros pacientes, se continúa con el diseño del adecuado estudio de investigación y finaliza con la aplicación de sus resultados a dichos pacientes. Es indudable que esta conexión directa entre actividad investigadora y práctica clínica genera una notable satisfacción al médico-investigador que las lleva a cabo, al comprobar cómo se puede mejorar la calidad de vida de nuestros pacientes, que es, al fin y al cabo, nuestro objetivo prioritario como médicos y como investigadores.
Conflicto de interesesJ.P. Gisbert: asesoramiento científico, soporte para investigación o actividades formativas: MSD, Abbvie, Hospira, Pfizer, Kern Pharma, Biogen, Takeda, Janssen, Roche, Sandoz, Celgene, Ferring, Faes Farma, Shire Pharmaceuticals, Dr. Falk Pharma, Tillotts Pharma, Chiesi, Casen Fleet, Gebro Pharma, Otsuka Pharmaceutical, Vifor Pharma, Mayoly, Allergan, Diasorin.
M. Chaparro: asesoramiento científico, soporte para investigación o actividades formativas: MSD, Abbvie, Hospira, Pfizer, Takeda, Janssen, Ferring, Shire Pharmaceuticals, Dr. Falk Pharma, Tillotts Pharma.

