




A high percentage of patients with familial adenomatous polyposis (FAP) have extraintestinal manifestations. However, FAP associated with pancreatic cancer, such as in the case described below, is uncommon.
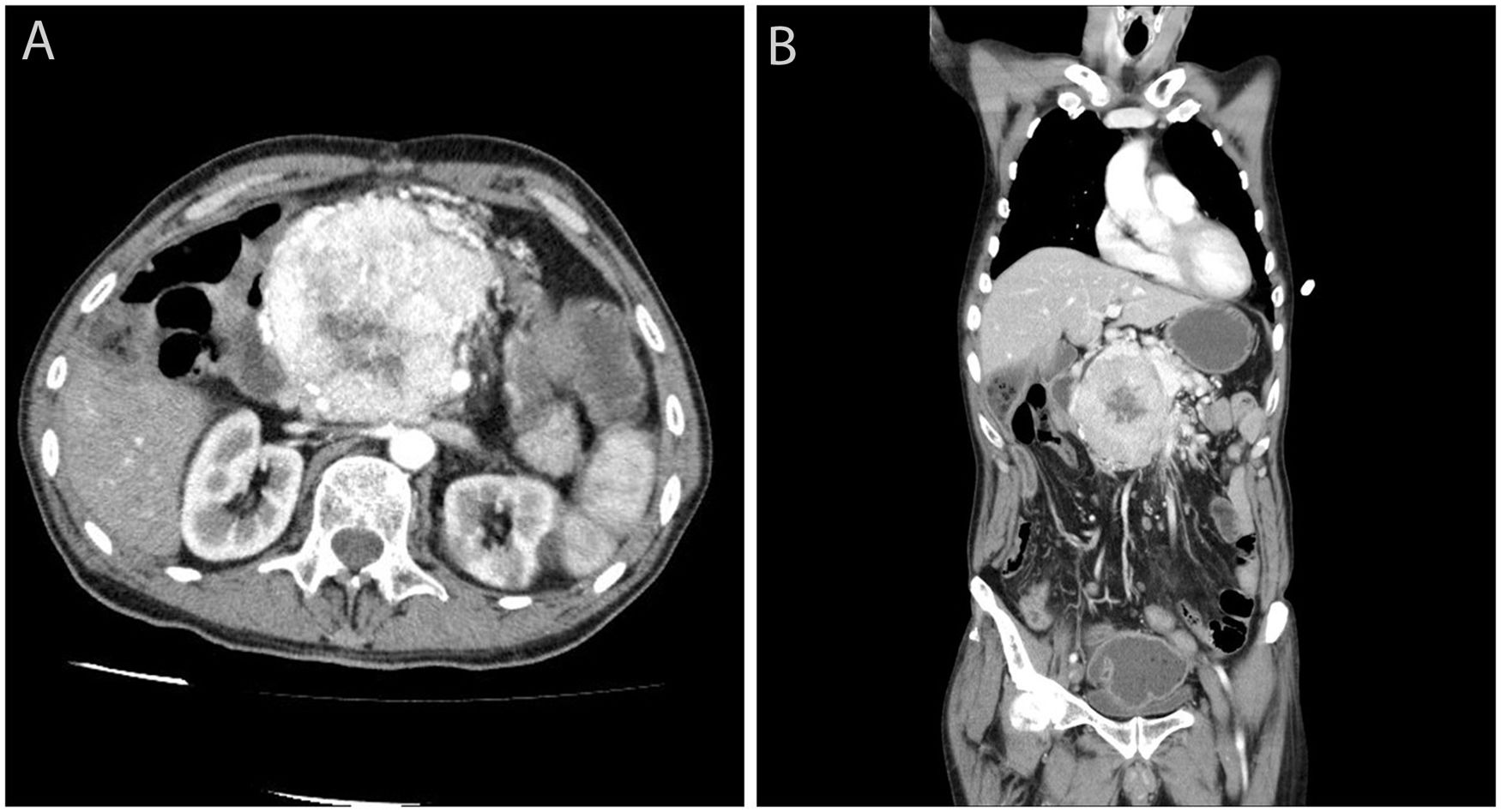
We present the case of a 65-year-old male former smoker with a history of Arnold-Chiari type I malformation, partial ulcer-related gastrectomy and FAP who, in his youth, had undergone total proctocolectomy and ileoanal anastomosis with ileal pouch and endoscopic resection of duodenal adenomas. He had stopped attending his medical check-ups in recent years due to depression. The patient consulted with epigastric pain and vomiting. On examination, a hard mass was palpable in the epigastrium, and ultrasound confirmed the presence of a solid, heterogeneous tumour adjacent to the pancreas. The CT scan showed a voluminous, hypervascularised 9×8cm mass, with an area of central necrosis, at the head of the pancreas compressing the duodenum and neighbouring vascular structures (Fig. 1). No liver metastases were detected. Chromogranin levels were found to be elevated (209.4ng/ml). Octreotide scintigraphy identified intense activity in the region of the pancreatic tumour, but no uptake of the marker in other locations. Ultrasound-guided percutaneous biopsy of the lesion confirmed a well differentiated, chromogranin- and synaptophysin-positive, intermediate-grade (WHO G2) neuroendocrine tumour, with a Ki-67 index of less than 20%. The multidisciplinary team decided to operate on the lesion, but during surgery it was discovered to be unresectable. The patient was started on chemotherapy (streptozocin/5-fluorouracil), but died a few months later as a result of pneumonia with sepsis.
 Computed tomography transverse view showing a hypervascularised tumour with central necrosis at the head of the pancreas, suggestive of a pancreatic neuroendocrine tumour. (B) Computed tomography coronal view.")
FAP is the most common inherited polyposis syndrome. Even so, it only affects one in every 10,000–20,000 people. It is characterised by multiple adenomatous polyps in the colon, from tens to thousands in number. FAP occurs due to a germline mutation in the APC gene, and is transmitted with an autosomal dominant inheritance pattern. Up to 70% of individuals with FAP develop extraintestinal manifestations, which include desmoid tumours, osteomas, epidermoid cysts, dental abnormalities, congenital hypertrophy of the retinal epithelium, duodenal and periampullary adenomas, papillary thyroid carcinoma and hepatoblastoma. It is known that the location of the mutation within the APC gene determines the clinical phenotype, the severity, the risk of cancer and the likelihood of certain extraintestinal manifestations, depending on the codons affected.
Pancreatic tumours are uncommon in individuals with FAP. However, there are reports in the medical literature of cases of FAP and pancreatic tumours of different types (exocrine, endocrine and stromal). Nevertheless, whether or not there is a common genetic association responsible for both conditions remains the subject of debate. Patients with FAP are estimated to have a four-fold higher risk of pancreatic carcinoma than the general population, with the total risk being around 2% over their lifetime.1 Available data are limited and it remains uncertain whether or not the germline mutation of the APC gene is responsible for the development of pancreatic cancer. Isolated cases of FAP have also been documented with other atypical pancreatic tumours such as pseudopapillary tumour, acinar cell carcinoma, pancreatoblastoma and neuroendocrine tumours (NET).2,3 There are reports of FAP and NET in other locations, e.g. gastric or duodenal or metastases of NET of unknown origin.4 The incidence of NET of the pancreas is 2–4 cases/100,000 population, accounting for less than 1% of all pancreatic tumours. Few reports in the literature describe cases of FAP in association with NET of the pancreas.5 There is no known genetic abnormality at present which explains this association.
As FAP and pancreatic NET are both very rare conditions, we suggest that this association may not be accidental and that the germline mutation of the APC gene may also be responsible for the development of NET and other pancreatic tumours in individuals with FAP, through a common genetic pathway not yet identified. It would be interesting to be able to gather a larger series of cases to carry out an in-depth genetic study and determine whether or not there are solid links between these tumours.
Please cite this article as: Pardillos Tomé A, Bajador Andreu E, Comín Orce A, Marcilla Córdoba F. Poliposis adenomatosa familiar asociada a tumor neuroendocrino de páncreas. Gastroenterol Hepatol. 2021;44:130–131.
