




Epithelioid sarcoma (ES) is a rare and aggressive soft tissue malignant tumour that most often originates under the skin of the hands, forearms, feet or lower legs.1,2 It mainly affects adolescents and young adults. It often metastasises to the lung and treatment is surgical, although there is no clear line of treatment in the case of regional or distant metastases.1–3 We present a case of primary jejunal ES.
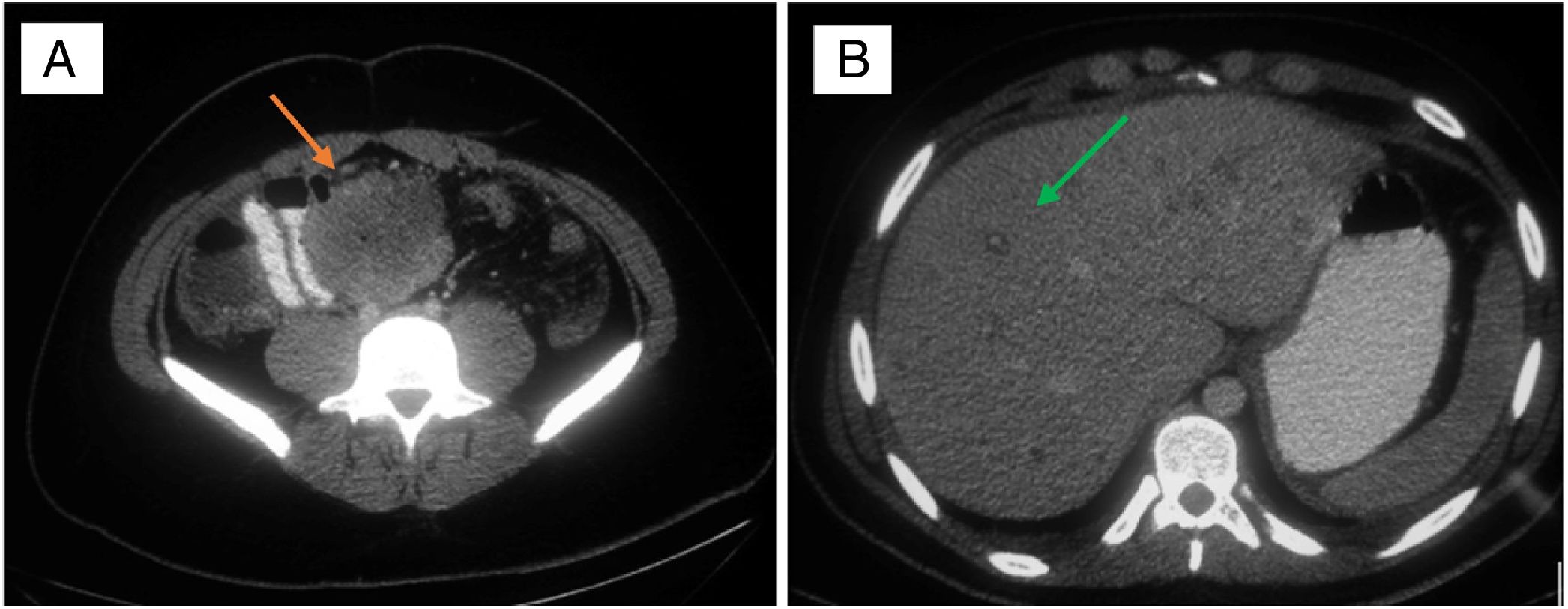
In a 19-year-old male patient, with no relevant history, with diffuse abdominal pain for two months, worse over the last 24–48 h, associated with a weight loss of 5 kg. On examination, he had signs of diffuse peritonism. Laboratory tests showed: leucocytes 15,700 mg/dl; prothrombin activity 42.6%; GGT 108 U/l; and CRP 118 mg/dl. CT showed an abscessed mass of intestinal origin measuring 7.6 × 6.5 cm, multiple predominantly peripheral liver lesions, and abundant ascites (Fig. 1). An exploratory laparotomy discovered 1.5 L of ascites fluid, a perforated tumour in the mid jejunum with localised purulent peritonitis, peritoneal carcinomatosis, and multiple liver metastases. Segmental resection of the jejunum was performed, including the tumour mass, and samples were taken of the ascites fluid, peritoneal implants and liver lesions. The patient made good postoperative progress and was discharged on day eight. He was readmitted 72 h later due to ascites refractory to medical treatment and required paracentesis to evacuate the fluid. Follow-up CT at 15 days showed a significant increase in liver and peritoneal lesions and massive ascites, leading to progressive worsening of the patient's condition and he died 27 days after the intervention. The histological study reported an intestinal wall with mucosal ulceration and multiple peritoneal implants due to proximal epithelioid sarcoma with rhabdoid, spindle-shaped and clear cell regions with a mitotic rate of 38/50 HPF and proliferative index of 40%. No lymphovascular or perineural invasion. Chronic lymphadenitis in the 33 isolated nodes. The immunohistochemical profile showed positivity for cytokeratin AE1/AE3, cytokeratin 19, WT-1, vimentin and CD34 and negativity for ALK, S-100, melan-A, HMB-45, muscle actin, desmin, MyoD1, myogenin, CD45, CD30, CD56, CD117, CD10, calretinin, CD15, CD99, cytokeratins 7, 20 and 5/6, CD31, D2-40, Ber-EP4, factor VIII and oestrogen receptors.
 Abscessed mass of intestinal origin (red arrow); B) Liver metastases (green arrow).")
Described by Enzinger in 1970, ES is a rare mesenchymal neoplasm.1–5 There are two variants: classic and proximal. Both types are associated with loss of SMARCB1/INI1 protein expression.4 The classic type most commonly affects the distal upper limbs in adolescents and young adults, with a preponderance in males.1,2 It presents as firm, painless, superficial nodules that give rise to chronic non-healing ulcers with elevated margins, and can be multifocal. They can also develop in the tenosynovial tissue and spread along the nerves and fascial planes. It is commonly multifocal, with local recurrence, and regional and distant metastases (40–57%).2,3 These are aggressive tumours and surgery is the treatment of choice. The prognosis is poor, particularly in the case of metastatic disease. The proximal type is found primarily in the pelvic, perineal and genital regions of young and middle-aged adults. This subtype is less common, but more aggressive.4 Five-year survival is estimated in 32–78% of cases.3,5 There is no evidence of any beneficial effect of lymphadenectomy associated with resection of the lesion on the local or distant recurrence rate. When surgery and radiotherapy are combined, it appears that there is better local control of the disease, but 40–60% of patients will die from metastatic disease.3–5
In various recently published reviews on ES, 77% of patients recurred and 45% developed metastases, predominantly in the lung (51%), regional lymph nodes (34%), scalp (22%), bone, brain, liver and pleura.3,4 Factors influencing a less unfavourable outcome are young age at diagnosis, being female and tumour size <5 cm.3–5 Older age, local multifocal disease, proximal location, axial location, depth of invasion, high mitotic activity, necrosis, vascular invasion, tumour bleeding, rhabdoid cytomorphology and inadequate excision are predictors of poor survival free from distant metastasis.3–5 ES of intestinal origin is extremely rare, with very few cases reported in the literature.3,5
FundingNo funding was received for this work.
Conflicts of interestThe authors declare that they have no conflicts of interest.
Please cite this article as: Serradilla-Martín M, Palomares-Cano A, Carrillo-Colmenero AM, Ramírez-Tortosa CL. Sarcoma epitelioide de origen intestinal. Gastroenterol Hepatol. 2021;44:298–299.
