




Amyloidosis is a rare disease characterized by deposition of insoluble, fibril-forming amyloid proteins in the extracellular space of organs, eventually producing insufficiency and end-organ dysfunction. The most common form of amyloidosis is light-chain (AL) amyloidosis. AL amyloidosis is a clonal plasma cell disorder, being the kidney, heart and peripheral nerves the most commonly organs affected.1–4 Liver is often involved histologically (60–92%) in patients with AL amyloidosis, but most cases are clinically asymptomatic.1,4,5
This research presents the case of a 70-year-old man, without relevant past medical history, admitted to our hospital with progressive jaundice, choluria and abdominal distension for 2 weeks. There was no history of weight loss, encephalopathy or gastrointestinal bleeding. On physical examination, the patient presented generalized jaundice, mild peripheral edema and hepatomegaly, with a palpable liver until 3cm below the right costal margin. Laboratory data showed hyperbilirubinemia (7.5mg/dl) and cholestasis enzymes elevation (alkaline phosphatase 1273U/L; gamma-glutamyl transferase 705U/L). Hemogram, coagulation and renal function were both normal. Ultrasonography and computed tomography (CT) of the abdomen revealed hepatomegaly and portal hypertension signs, such as paraumbilical vein recanalization and mild quantity of ascites. Serologies for hepatitis A, B, C and E viruses, CMV, EBV and HIV were negative. Anti-nuclear, anti-mitochondrial and anti-smooth muscle antibodies were negative. Serum immunoglobulins (IgG, IgA, IgM) were negative. Proteinogram (alpha-1 globulin, alpha-2 globulin, beta globulin), ceruloplasmin and alpha 1-antitrypsin were in normal range.
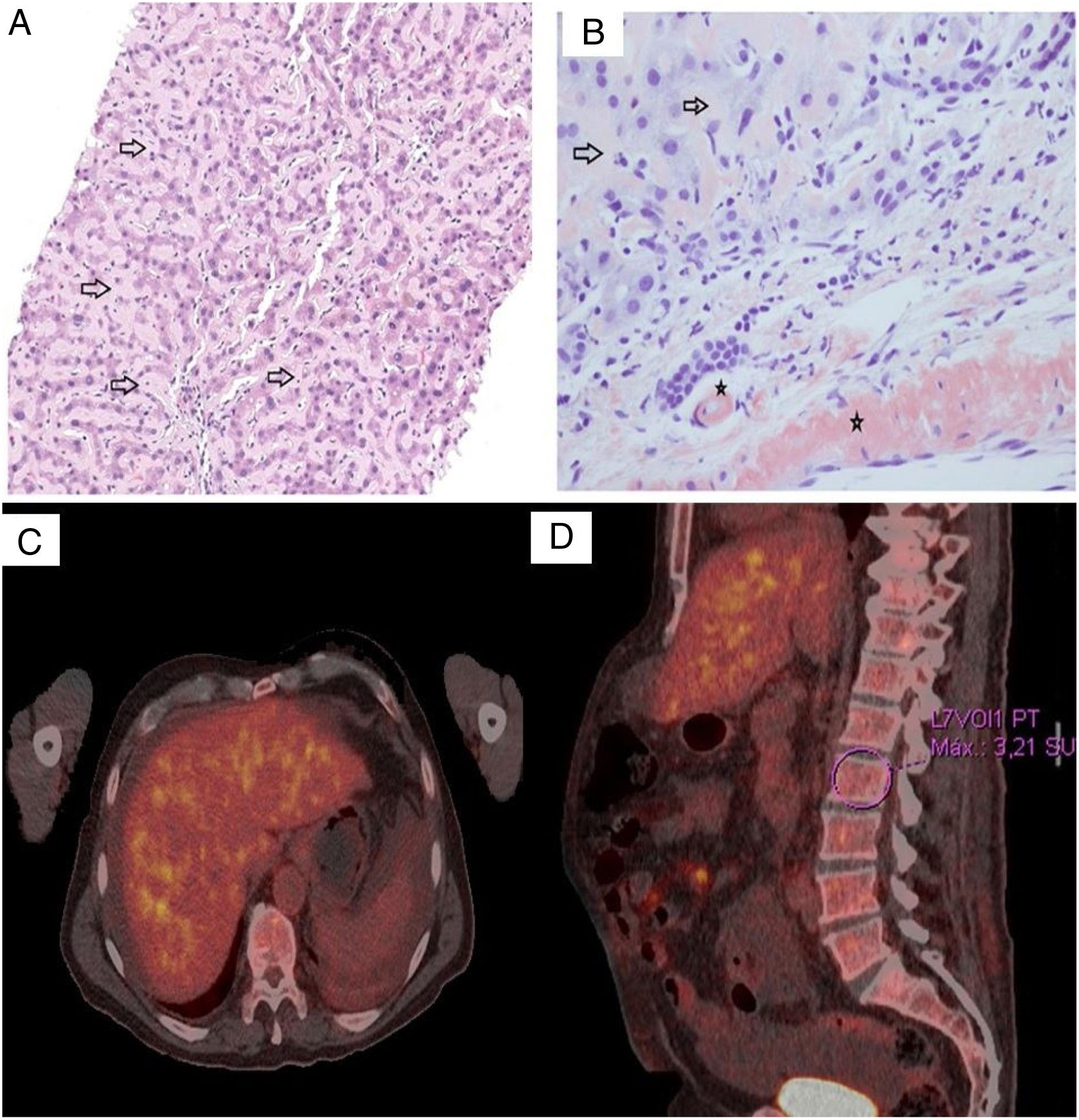
Due to the existence of an acute cholestatic hepatitis with non-identified etiological agent, we performed a percutaneous liver biopsy. The liver tissue biopsy showed perisinusoidal deposition of an eosinophilic amorphous substance (Fig. 1A). Examination under polarized light of sections stained with Congo red demonstrated apple-green birefringence (Fig. 1B). Immunohistochemistry staining was negative for amyloid A and λ light chain, and positive for κ light chain. The histopathological diagnosis was an AL amyloidosis with κ light chain deposits.
 Liver cell trabeculae are compressed by marked amyloid deposition in the sinusoids. Abundant acellular pink material (black arrows) on a H&E stain is seen in this high magnification picture. (B) The presence of amyloid was confirmed by apple green birefringence on Congo Red staining with polarized microscopy. The deposit was observed both in the sinusoids (arrows) as well as in the vessels wall (black star). (C, D) FDG-PET showed signs of hepatic infiltration (SUVmax=4.3) and axial skeleton bone marrow heterogeneous hypercaptation (SUVmax L2=3.2).")
(A) Liver cell trabeculae are compressed by marked amyloid deposition in the sinusoids. Abundant acellular pink material (black arrows) on a H&E stain is seen in this high magnification picture. (B) The presence of amyloid was confirmed by apple green birefringence on Congo Red staining with polarized microscopy. The deposit was observed both in the sinusoids (arrows) as well as in the vessels wall (black star). (C, D) FDG-PET showed signs of hepatic infiltration (SUVmax=4.3) and axial skeleton bone marrow heterogeneous hypercaptation (SUVmax L2=3.2).
The hematological study showed evidence of a monoclonal plasma cell proliferative disorder, appreciating in the laboratory data the presence of a serum and urine M protein, abnormal serum free light chain and serum protein electrophoresis (SPEP) IgG kappa. Clonal plasma cells could also be identified in the bone marrow. We observed 2.4% plasma cells with aberrant immunophenotype, so multiple myeloma presence was ruled out.
Bence Jones protein was detected in urine, yet kidney involvement was not found. Transthoracic echocardiogram (TTE) and electrocardiogram do not show cardiac involvement. Neurophysiological study was carried out, without appreciating signs of polyneuropathy or myopathy. Positron emission tomography with 18F-fluorodeoxyglucose (FDG-PET) was then performed to complete the study, revealing the presence of signs of hepatic infiltration and axial skeleton bone marrow heterogeneous hypercaptation (Fig. 1C, D).
While making the assessment, liver function worsened. The patient experienced a deterioration of the liver biochemistry, with bilirubin 24.1mg/dl and ALP 1065U/L, and developed moderate ascites, with good response to diuretic treatment. Signs of acute liver failure were not seen during assessment.
Therefore, with the diagnosis of AL amyloidosis with exclusively liver involvement, chemotherapy treatment was initiated with bortezomib (adjusted-dose 0.7mg/m2, according to data sheet because of hyperbilirubinemia), cyclophosphamide and dexamethasone. Clinical and laboratory improvement was initially achieved, bringing bilirubin levels down to 10mg/dl four weeks after initiation of treatment.
However, one month later the patient was readmitted to hospital due to hepatic decompensation with ascites, spontaneous bacterial peritonitis and bilateral pneumonia and finally died due to respiratory failure.
The clinical spectrum of hepatic amyloidosis can range from hepatomegaly (most frequently physical sign observed) and mild abnormal liver function tests, to more severe symptoms rarely observed, such as jaundice, intrahepatic cholestasis, portal hypertension, hepatic failure or spontaneous liver rupture.1,3–5 Jaundice and intrahepatic cholestasis as the primary manifestation of the disease is very uncommon (<5% cases).4
Hyperbilirubinemia and marked elevation of serum alkaline phosphatase may indicate a poor prognosis of hepatic amyloidosis.1,3–5
Diagnosis of hepatic amyloidosis is based on a high clinical suspicion, exclusion of other infiltrative disorders of the liver (tuberculosis, sarcoidosis, malignancy and glycogen storage diseases) and tissue biopsy stained with Congo red demonstrating amyloid deposits with apple-green birefringence.1,2,4,5
Treatment of AL amyloidosis is based on chemotherapy to eradicate the underlying clone, usually combinations of bortezomib, cyclophosphamide and dexamethasone.2,4 Patient who meet the criteria will be eligible for autologous hematopoietic cell transplantation.
Liver involvement in patients with amyloidosis is often an indicator of poor prognosis,3 having been reported as 9 months the median survival of patients with primary hepatic amyloidosis.1,3–5
Conflicts of interestThere are no financial or other conflicts of interest regarding this article.
