




El 31 de diciembre de 2019 se reportó el primer caso de COVID-19 en Wuhan, China, y desde entonces ha habido un interés creciente y sin precedentes por conocer todos los aspectos vinculados con esta nueva enfermedad. Uno de los temas que ha generado debate se vincula con la asociación entre la terapia antihipertensiva con inhibidores del sistema renina-angiotensina-aldosterona y la infección por el virus SARS-CoV-2. Si bien muchas preguntas siguen hoy en día sin poder ser respondidas, la intención de este comunicado es informar a los profesionales de la salud acerca del estado actual de conocimiento. Dado que este es un tema en constante evolución, se recomienda su actualización a medida que se presenten nuevas evidencias. A continuación daremos revisión a los estudios preclínicos y clínicos que relacionan el coronavirus con el sistema renina-angiotensina-aldosterona.
The first case of COVID-19 was reported on 31 December 2019 in Wuhan, China. Ever since there has been unprecedented and growing interest in learning about all aspects of this new disease. Debate has been generated as to the association between antihypertensive therapy with renin-angiotensin-aldosterone system (RAAS) inhibitors and SARS-CoV-2 infection. While many questions as yet remain unanswered, the aim of this report is to inform health professionals about the current state of knowledge. Because this is an ever-evolving topic, the recommendation is that it be updated as new evidence becomes available. Below, we provide a review of pre-clinical and clinical studies that link coronavirus to the RAAS.
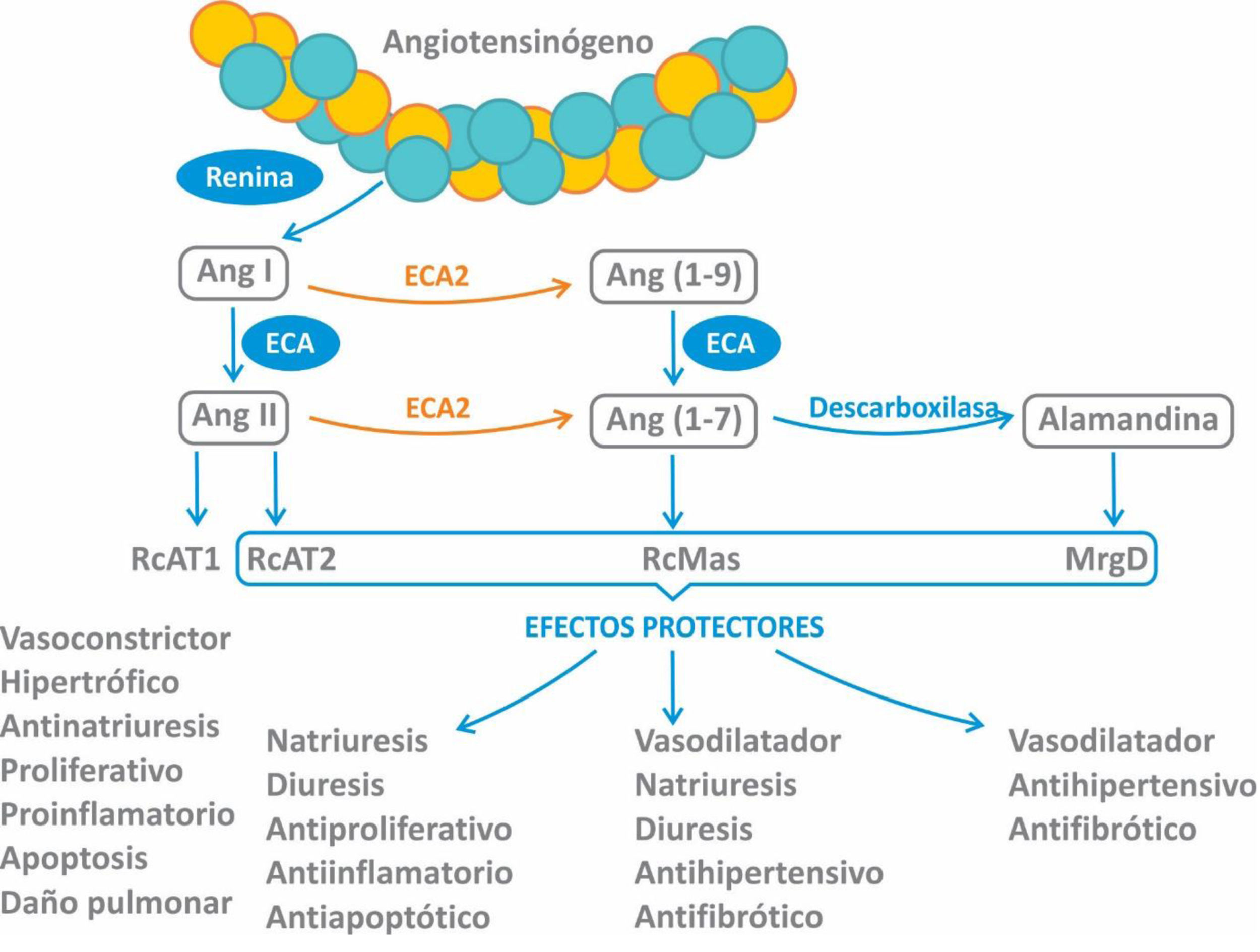
Se ha demostrado que la vía de entrada para el virus SARS-CoV-2 es la proteína enzimática transmembrana enzima convertidora de angiotensina tipo 2 (ECA2), que se encuentra en diferentes tejidos como el miocardio, el riñón, las vías respiratorias y a nivel vascular1. A nivel respiratorio la principal célula diana para el virus es la célula alveolar tipo ii o neumocito tipo ii, que representa el 5% de la superficie alveolar. Este tipo celular es el responsable de secretar el surfactante y es progenitor de los neumocitos tipo i en caso de lesión tisular2. A diferencia de la clásica ECA, la ECA2 degrada la angiotensina ii(Ang II), uniéndose a ella con una afinidad 400 veces mayor que a la Ang I (fig. 1)3. Cabe aclarar que, a pesar de compartir un 60% de homología estructural con la ECA, sus sitios catalíticos son diferentes, por lo que los fármacos inhibidores de la ECA (IECA) no inhiben la ECA24.
 y ECA2 en la cascada del sistema renina angiotensina. MrgD: receptor Mas acoplado a proteína G tipo D; RcAT1: receptor de Ang II tipo 1; RcAT2: receptor de Ang II tipo 2; RcMas: receptor Mas de Ang 1-7.")
La ECA2 es una proteína transmembrana con actividad de monocarboxipeptidasa que regula negativamente el sistema renina-angiotensina-aldosterona (SRAA) al convertir Ang II en Ang 1-7. También degrada Ang I para generar Ang 1-9 (sin actividad biológica conocida) y otros péptidos como desArg-bradiquinina, neurotensina 1-13, quinetensina, apelin-13 y dinorfina A 1-134. La conversión de Ang II a Ang 1-7 por parte de la ECA2 trae como consecuencia la disminución del efecto vasoconstrictor a través de 2 acciones principales5:
- -
Reduciendo los niveles del efector primario del SRAA, Ang II, y por ende la vasoconstricción.
- -
Aumentando la formación de Ang 1-7 con acciones vasodilatadoras a través de su unión al receptor Mas.
La ECA2 en su forma completa se encuentra anclada en la membrana plasmática, mientras que su forma acortada (o soluble) circula en bajos niveles en la sangre6. Existe un gran interés en dilucidar la función de la ECA2 en la enfermedad cardiovascular, tanto de la enzima transmembrana como también de la forma soluble.
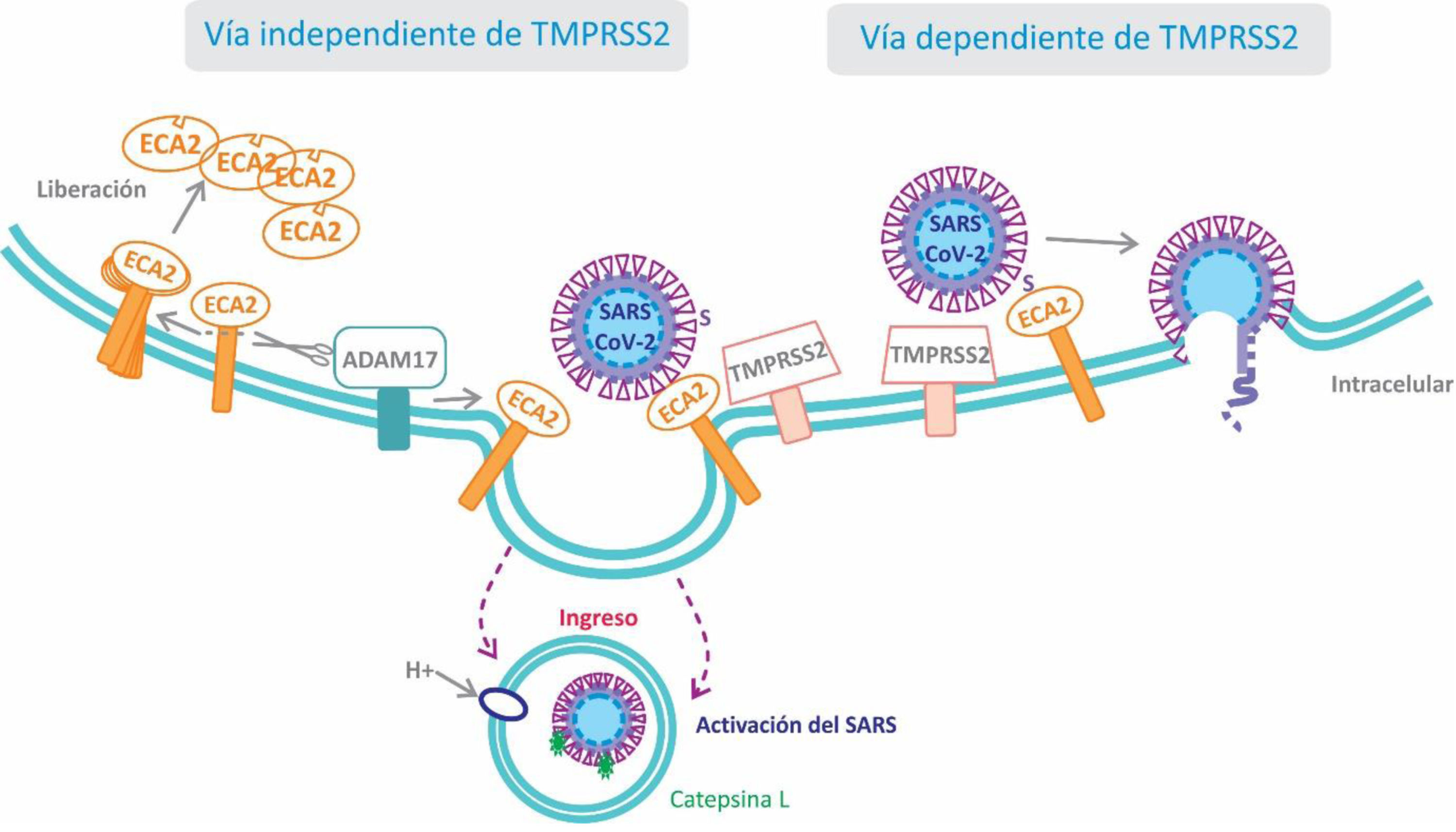
Para ingresar penetrar en las células el coronavirus interactúa, utilizando como receptor, con la ECA2 y serina-proteasas transmembrana de tipo ii (TMPRSS2) ubicadas en la superficie celular del huésped7. El virus, a través de su proteína S1 (Spike o pico), se une al dominio catalítico de la ECA2 con alta afinidad, incluso 10 a 20 veces más que el SARS-CoV8. Estudios sobre el comportamiento de su congénere SARS-CoV permitieron conocer el mecanismo de la infección celular. La unión del virus a la ECA2 desencadena un cambio conformacional en la proteína S del coronavirus, permitiendo la degradación proteolítica por las serina-proteasas TMPRSS2, exponiendo una subunidad de la proteína S que permite su fusión a la membrana celular y facilita el ingreso del virus en el interior de la célula huésped9. Por lo enunciado, las TMPRSS2 serían también un potencial objetivo terapéutico para disminuir la infección viral (fig. 2).

Existe una vía alternativa de entrada del virus a la célula huésped que no implica la activación de las TMPRSS2 (vía independiente de TMPRSS2). En este caso, tras la unión del virus a la ECA2 se forma un endosoma dentro del cual la proteína S es clivada y activada por la cisteína-proteasa catepsina L dependiente de pH9.
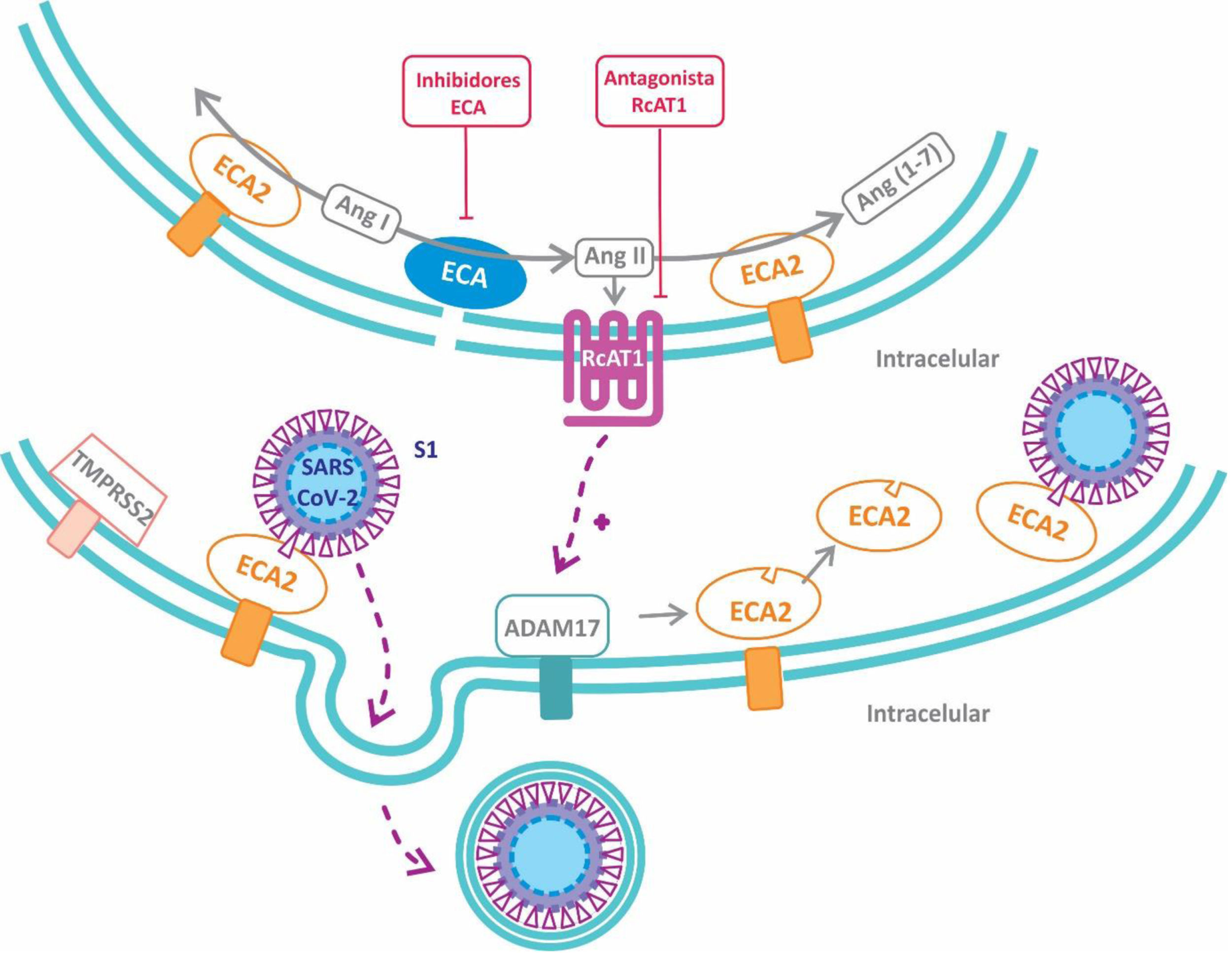
Por otro lado, resulta de especial interés la participación de la proteína transmembrana disintegrina y metaloproteasa 17 (ADAM17), que es activada por la proteína S viral y cliva a la ECA2 produciendo su eliminación de la superficie celular10. Normalmente la ECA2 protege del daño pulmonar ocasionado por la Ang II al degradarla a Ang 1-711. Debido a que se ha informado de que los niveles de ECA2 durante una infección por SARS-CoV se encuentran disminuidos, la proteína ADAM17 desempeñaría un rol esencial en el daño pulmonar al promover la eliminación de la ECA212. La observación de que los pacientes con COVID-19 presentan niveles bajos de potasio en sangre, permite especular que la disminución de la ECA2 llevaría a un incremento de los niveles de Ang II, y por ende de aldosterona, con la consecuente pérdida de potasio13. Consistentemente, ha sido reportado recientemente en pacientes con COVID-19 que los niveles plasmáticos de Ang II se correlacionan positivamente con la carga viral de SARS-CoV214. Adicionalmente, el receptor AT1 de Ang II regula al alza la proteína ADAM17, incrementando así los niveles circulantes de la fracción soluble de ECA2 (fig. 3)7.
Relación entre coronavirus tipo 2 del síndrome respiratorio agudo grave e hipertensión arterial: hipótesis postuladas en la formación de la forma soluble de enzima convertidora de angiotensina 2 (ECA2). RcAT1: receptor de Ang II tipo 1; TMPRSS2: serina-proteasas transmembrana de tipo ii.")
Se han postulado diversas hipótesis para explicar una mayor severidad de la enfermedad del SARS-CoV-2 en pacientes hipertensos:
1) Una de estas hipótesis postula que la mayor expresión de ECA2 en un tejido dado, como el pulmón, incrementaría la infectividad del virus y empeoraría el curso de la enfermedad. Esto fue demostrado en cultivos celulares en donde la mayor expresión de ECA2 se asociaba a una mayor tasa de infección por el virus15. Resulta interesante la observación de que las personas mayores y los hombres presentan peor evolución de la enfermedad y mayores tasas de mortalidad por COVID-1916. Esto plantearía el interrogante de cómo varían los niveles de ECA2 en el pulmón con la edad y el género. A nivel experimental se han demostrado mayores niveles de expresión pulmonar de ECA2 en ratas Sprague Dawley jóvenes comparadas con ratas envejecidas, contribuyendo a una mayor tasa de infección en las jóvenes17. Este último hallazgo nos permitiría inferir que la mayor infectividad en personas mayores no se relaciona con los niveles de expresión de ECA2, aunque deberían evaluarse otras variables. Por otra parte, las evidencias acerca de diferencias en la expresión de ECA2 plasmática según sexo y edad en seres humanos son controvertidas. Un estudio reporta que no existen diferencias en la expresión de ECA2 plasmática entre hombres y mujeres, aunque sí mayores niveles de expresión en mujeres mayores que en jóvenes18. En oposición, en otro trabajo se ha determinado que una mayor expresión de ECA2 plasmática se asocia a hombres de mayor edad19. En conclusión, hasta el momento, ningún estudio clínico ha demostrado fehacientemente la validez de esta hipótesis.
2) Otra hipótesis relaciona la infección del SARS-CoV-2 con ciertos agentes antihipertensivos, como los inhibidores del SRAA. Existen hallazgos controvertidos al respecto. Algunos estudios experimentales demuestran que efectivamente los IECA (lisinopril y enalapril) y los antagonistas del receptor de Ang II, ARA-II (losartán y telmisartán) pueden aumentar los niveles de expresión de ECA220–22. No obstante, otros trabajos demuestran lo contrario23,24, o incluso la no asociación. Adicionalmente, unos pocos estudios clínicos demostraron que no existía ninguna asociación entre los niveles circulantes de la fracción soluble de ECA2 y el uso de IECA y ARA-II19,25. Dado que el tratamiento con IECA o ARA-II tiene un impacto diferente en varios componentes del SRAA, ya sea directamente o mediante bucles de retroalimentación, se ha postulado la posibilidad de que ambos grupos de inhibidores del SRAA tengan un efecto diferencial en pacientes con COVID-19. Sin embargo, faltan estudios que demuestren fehacientemente un efecto de los IECA o ARA-II sobre la expresión o actividad de ECA2 en los pulmones en humanos. Los estudios clínicos llevados a cabo hasta el día de hoy no han demostrado que existan diferencias entre ambos tratamientos en términos de aumento del riesgo de infección por SARS-CoV-2, o de desarrollo de resultados graves en pacientes con COVID-1926–30. No obstante, es de destacar que la mayoría de estos estudios son observacionales y retrospectivos, siendo necesarios datos a partir de estudios controlados y aleatorizados para confirmar estos hallazgos.
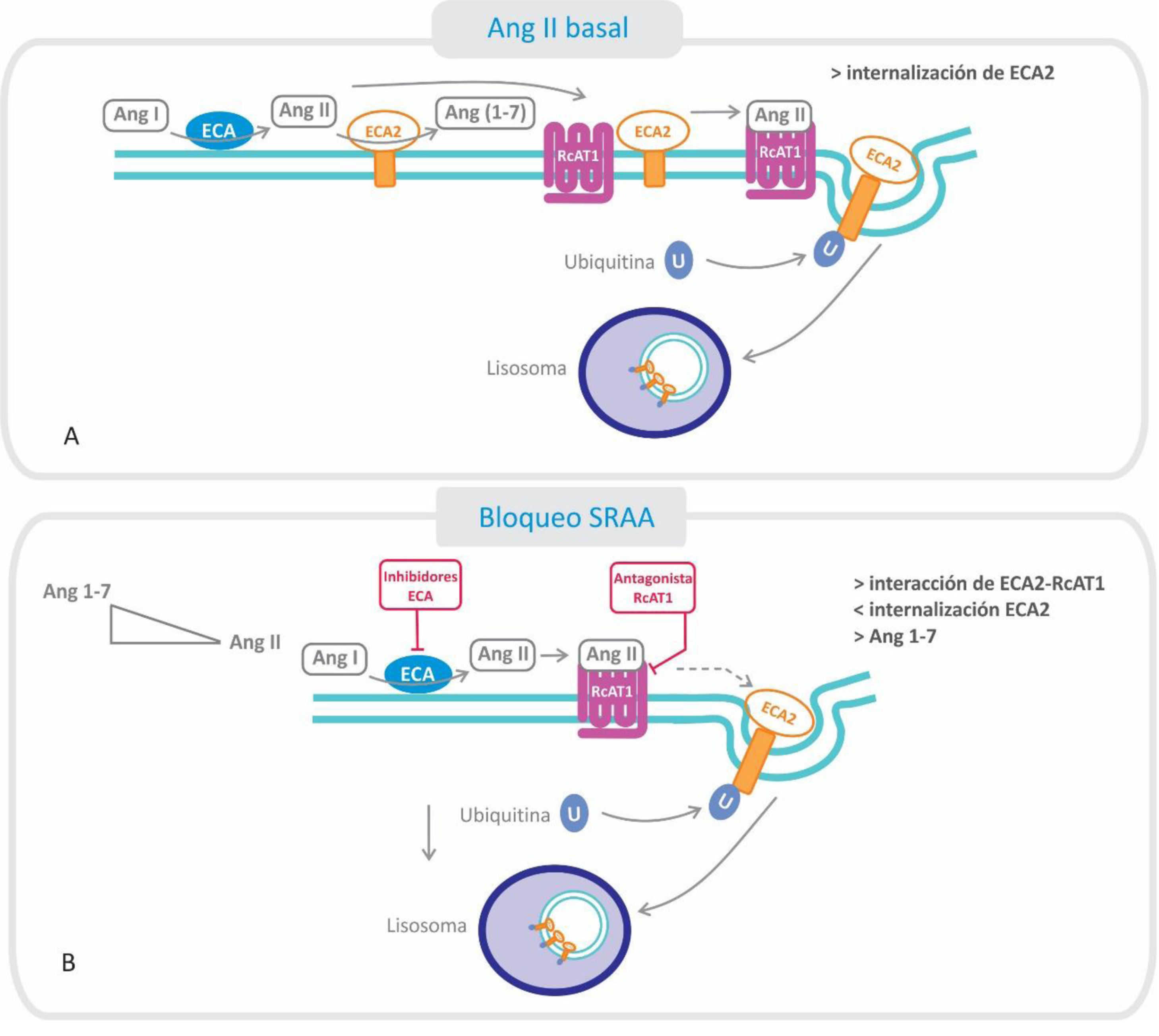
3) La tercera hipótesis postula que el uso de los IECA y ARA-II conllevaría un beneficio en relación con la infección y gravedad por COVID-19. Se ha demostrado que la ECA2 interactúa con el receptor AT1 formando un complejo que dificultaría la interacción de la ECA2 con la proteína S viral. La Ang II, al unirse al receptor AT1, disminuye los niveles de expresión de ECA2 al promover su internalización en lisosomas (fig. 4)31De esta manera, el uso de IECA o ARA-II evitaría la disociación del complejo ECA2/AT1 al prevenir la internalización y degradación de ECA231. Esto trae como consecuencia, por un lado, la persistencia del complejo ECA2-AT1 que dificulta la interacción entre el virus y la ECA2, y por otro lado, que la ECA2 pueda degradar la Ang II a Ang 1-7, disminuyendo así los efectos deletéreos de la Ang II sobre el pulmón. En términos de beneficios en la evolución de la enfermedad por COVID-19, el estudio de Mehra et al. ha mostrado que los IECA, a diferencia de los ARA-II, se asocian con una mayor probabilidad de supervivencia al alta hospitalaria32
 y la enzima convertidora de angiotensina 2 (ECA2): efectos del bloqueo del sistema renina-angiotensina-aldosterona. A. Estado basal: la Ang II formada se une al RcAT1, disminuyendo la interacción de este con la ECA2, fenómeno que favorece la endocitosis y posterior degradación, disminuyendo el nivel de ECA2 en la membrana. B. Cuando se utilizan inhibidores de la ECA o antagonistas de los RcAT1, ARA-II, disminuye la cantidad de Ang II formada y aumenta su degradación (por ECA2), estabilizándose la interacción del RcAT1 y la ECA2. Por consiguiente, disminuye la endocitosis de ECA2 y se favorece la conversión de Ang II a Ang 1-7.")
Relación entre el receptor AT1 (RcAT1) y la enzima convertidora de angiotensina 2 (ECA2): efectos del bloqueo del sistema renina-angiotensina-aldosterona.
A. Estado basal: la Ang II formada se une al RcAT1, disminuyendo la interacción de este con la ECA2, fenómeno que favorece la endocitosis y posterior degradación, disminuyendo el nivel de ECA2 en la membrana.
B. Cuando se utilizan inhibidores de la ECA o antagonistas de los RcAT1, ARA-II, disminuye la cantidad de Ang II formada y aumenta su degradación (por ECA2), estabilizándose la interacción del RcAT1 y la ECA2. Por consiguiente, disminuye la endocitosis de ECA2 y se favorece la conversión de Ang II a Ang 1-7.
4) Otra hipótesis posible plantea la importancia que tiene la ECA2 para prevenir el daño pulmonar asociado a la infección viral. Altos niveles de Ang II a nivel pulmonar se asocian con un mayor daño e injuria tisular por su efecto proinflamatorio y prooxidante, mientras que la presencia de Ang 1-7, a través de la activación del receptor Mas, tendría el efecto contrario. Se demostró de esta manera que ratones knockout para la ECA2 presentaban mayor severidad y mortalidad por neumonía viral. En este sentido, la presencia de la ECA2 sería crucial para disminuir los niveles de Ang II e incrementar los de Ang 1-712,33.
5) Basándonos en la evidencia mencionada Sommerstein et al. han postulado que los fármacos que inhiben el SRAA podrían ejercer un rol durante el curso severo de la COVID-19. Los autores plantean así una especie de arma de doble filo, dependiendo de la fase de la enfermedad en la que nos situemos: el aumento de la expresión basal de ECA2 podría aumentar potencialmente la infectividad, por lo que el uso de IECA y/o ARA-II sería un factor de riesgo, mientras que una vez establecida la infección existe una disminución de la ECA2, por lo que el uso de estos inhibidores del SRAA podría resultar beneficioso34.
Posibles estrategias terapéuticas a investigarComo posibles estrategias a futuro para combatir la enfermedad COVID-19 se encuentran, entre otras:
- -
Desarrollo de una vacuna basada en la proteína Spike S1.
- -
Producción de anticuerpos neutralizantes contra la proteína S1.
- -
Inhibición de la serina-proteasa transmembrana (TMPRSS2) (mesilato de camostato).
- -
Administración de la forma soluble de la ECA2.
- -
Bloqueo de la entrada del SARS-CoV-2 (Arbidol).
- -
Inhibición de la proteinquinasa asociada a AP2, proteína reguladora de la endocitosis viral (baricitinib).
- -
Bloqueo de la replicación SARS-CoV-2 por inhibición de ARN polimerasa viral (análogo nucleosídico remdesivir) o de la endonucleasa cap-dependiente (baloxavir marboxilato) o de la proteína básica 1 de polimerasa (favipiravir).
- -
Inhibición de proteasas virales necesarias para su replicación (darunavir; lopinavir/ritonavir).
- -
Bloqueo de la glicosilación del receptor ECA2 y de la producción de citoquinas inflamatorias (cloroquina e hidroxicloroquina).
- -
Bloqueo del receptor de IL-6 (tocilizumab).
- -
Inhibición de producción de citoquinas proinflamatorias (nitazoxanida).
Actualmente están en marcha estudios clínicos cuyo objetivo es evaluar las consecuencias y seguridad del bloqueo farmacológico del SRAA en la COVID-19:
- -
Administración de ECA2 humana recombinante vs. placebo: NCT04335136; EUCTR2020-001172-15-DK.
- -
Tratamiento con losartán vs. placebo: NCT04311177; NCT04312009.
- -
Tratamiento con telmisartán vs. placebo: NCT04360551.
- -
Tratamiento con valsartán vs. placebo: EUCTR2020-001320-34-NL; NCT04335786.
Si bien los datos disponibles de estudios en China y en Italia demuestran inequívocamente la asociación entre enfermedad cardiovascular, hipertensión y diabetes con una mayor mortalidad por COVID-1935–39, aún no está clara la relación entre la hipertensión arterial y el bloqueo del SRAA en la COVID-19. En humanos la evidencia actual proviene de estudios no ajustados o ajustados de forma incompleta, lo que implica sesgos de selección, de clasificación y presencia de variables que generan confusión (edad, duración y tipo de medicación antihipertensiva y carga de comorbilidad). Si bien existe evidencia in vitro de que el SARS-CoV-2 se une a los receptores ECA2, y que estos se encuentran aumentados en presencia de IECA o ARA-II, no hay evidencia en este momento de que la exposición a estos fármacos facilite la entrada del coronavirus, ni de que produzcan un mayor riesgo de COVID-19.
Los beneficios de IECA y ARA-II en la prevención primaria y secundaria de las enfermedades cardiovasculares son indudables, y han sido demostrados por numerosos ensayos clínicos controlados. Por ello, en función de la evidencia disponible hasta la fecha, no hay elementos que sugieran la necesidad del reemplazo ni de la suspensión de IECA o ARA-II en pacientes expuestos o con COVID-19 demostrada. Estos fármacos deben ser iniciados o mantenidos en pacientes con hipertensión, fallo cardíaco, infarto de miocardio y nefropatía diabética de acuerdo a las guías actuales, independientemente del estado de SAR-S-CoV-2. La suspensión y/o reemplazo del tratamiento en pacientes que reciban IECA o ARA-II no debe recomendarse como estrategia preventiva ni de tratamiento de la infección por SARS-CoV-2. A día de hoy las principales sociedades científicas (entre ellas la Sociedad Europea de Cardiología, la Sociedad Europea de Hipertensión, la Sociedad Internacional de Hipertensión, la Sociedad Española de Hipertensión y la Asociación Americana del Corazón, además de la propia Sociedad Argentina de Hipertensión Arterial) se han posicionado con indicaciones claras al respecto, recomendando continuar con el tratamiento de IECA o ARA-II, dada la falta de evidencia que apoye lo contrario40–45. Sin embargo, esta conclusión debe considerarse en constante revisión.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.