




Haemophagocytic lymphohistiocytosis (HLH) is characterised by T cell and macrophage activation and excessive production of inflammatory cytokines. Genetic diagnosis is required to discriminate between primary forms (familial HLH, FHL), due to mutations in genes involved in cytolysis, and secondary forms. We aimed to analyse the genes coding for Munc13-4 (UNC13D) and syntaxin-11 (STX11) proteins in search of mutations that might explain HLH in 5 patients without perforin defects.
Materials and methodsPerforin expression was evaluated by flow cytometry, sCD25 was measured by ELISA and NK activity was investigated by the conventional functional assay. Coding regions and exons surroundings were sequenced for PRF1, UNC13D and STX11 genes.
ResultsP1 and P2 developed severe early-onset HLH, P1 died at 6 months. P3, with a sister who died after HLH, responded well to treatment (HLH-2004), and had a second HLH episode two years later. P2 developed HLH at year 7 while in complete remission after lymphoblastic leukaemia. P4 and P5 were brothers who died at 5 and 6 years old due to an HLH and EBV mononucleosis infection. XLP was discarded because P4 was a girl. P1 and P3 showed mutations in UNC13D previously described as pathogenic. There were no changes in STX11.
ConclusionsUNC13D mutations were found in 50% of the HLH families without perforin defects and STX11 defects were not detected. These results agree with published series in which mutations in UNC13D explain up to 50% of FHL without PRF1 mutations, supporting a heterogeneous genetic background for this disease.
La linfohistiocitosis hemofagocítica (HLH) se caracteriza por la activación incontrolada de células T y macrófagos y producción excesiva de citoquinas inflamatorias. El diagnóstico genético es necesario para distinguir entre formas primarias (HLH familiar, FHL), debidas a mutaciones en genes implicados en citolisis, y secundarias. Nuestro objetivo es analizar la presencia de mutaciones en los genes que codifican para Munc13-4 (UNC13D) y sintaxina-11 (STX11) en cinco pacientes con HLH no asociado a defecto de perforina.
Materiales y métodosSe evaluó la expresión de perforina por citometría, CD25s por ELISA y la actividad NK con un ensayo funcional. Se secuenciaron los exones y regiones flanqueantes de los genes PRF1, UNC13D y STX11.
ResultadosP1 y P2 desarrollaron HLH severo de inicio temprano, P1 falleció con 6 meses. P3, con una hermana fallecida tras HLH, respondió adecuadamente al tratamiento (HLH-2004), presentando un segundo episodio dos años después. P2 desarrolló HLH a los 7 años de edad estando en remisión completa de una leucemia linfoblástica previa. P4 y P5 son hermanos que fallecieron con 5 y 6 años tras HLH y mononucleosis por infección EBV. Se descartó XLP ya que uno de los pacientes era niña. P1 y P3 presentaron mutaciones en UNC13D previamente descritas como patogénicas. No se encontraron alteraciones en STX11.
ConclusionesSe encontraron mutaciones en UNC13D en el 50% de las familias con HLH sin defectos de perforina y no se detectaron defectos en STX11. Este resultado concuerda con series publicadas en las que mutaciones en UNC13D explican hasta el 50% de la FLH sin mutaciones en PRF1, y apoyan una base genética muy heterogénea para esta enfermedad.
Haemophagocytic lymphohistiocytosis (HLH) is a severe condition characterised by the uncontrolled activation of polyclonal CD8+ T lymphocytes and macrophages. Phagocytosis of blood cells by macrophages (haemophagocytosis) is the hallmark of this syndrome. The activated cells infiltrate multiple organs, including bone marrow, spleen, lymph nodes, liver and brain, and produce considerable amounts of inflammatory cytokines such us γ-interferon, interleukin-6, interleukin-18 and tumor necrosis factor-α. This hipercytokinemia is presumably responsible for the necrosis and failure of the infiltrated organs, and for the onset of clinical and biological manifestations that characterise HLH. Main clinical features of this disorder include fever, hepatosplenomegaly, cytopenia and neurological abnormalities.1
HLH comprises a primary form and a secondary form that may be difficult to distinguish from each other. The primary, inherited form, named familial haemophagocytic lymphohistiocytosis (FHL), has an autosomal recessive inheritance and an incidence of 1:50,000 births. Secondary form may develop after a strong activating stimulus of the immune system, such as viral infections (frequently EBV) or malignancies. Demonstration of frequent association with common pathogens, together with evidence of impaired natural killer cytotoxic activity, provided the rationale for considering HLH as a selective immunodeficiency.2,3
According to the genetic causative defect, there are five FHL types (FHL1–FHL5).4 FHL1 maps on chromosome 9q21.3-q22. FHL2 to FHL5 types are due to mutations in genes coding for proteins involved in the granule-dependent cytotoxic function of lymphocytes. Deficiency in the cytolytic effector perforin, present in cytotoxic granules, was the first genetic defect identified as causing FHL (PRF1 gene, FHL2).5 Perforin is critical for the pore formation in the membrane of the target cell and ulterior entrance of granzymes for the initiation of death. Mutations in the gene coding for Munc13-4 (UNC13D gene) are the identified cause for FHL3.6 The exocytosis of cytotoxic granules in Munc13-4-deficient T and NK lymphocytes is impaired, as Munc13-4 is critical for the maturation of lytic granules and in its absence, the granules cannot release their content into the immunological synapse. More recently, mutations in genes coding for syntaxin 11 (STX11 gene) and Munc18-2 (STXBP2 gene) proteins involved in membrane fusion events have been found responsible for FHL4,7 and FHL5,8,9 respectively.
Last diagnostic HLH guidelines, published in 2007,10 indicate that the diagnosis of HLH can be established when (a) there is a molecular diagnosis consistent with HLH, and/or (b) five out of the following diagnostic criteria are fulfilled: (1) fever, (2) splenomegaly, (3) cytopenias (affecting at least 2 lineages in the peripheral blood), (4) hypertriglyceridemia and/or hypofibrinogenemia, (5) haemophagocytosis in bone marrow or spleen or lymph nodes without evidence of malignancy, (6) low or absent NK-cell activity, (7) ferritin ≥500μg/l and (8) soluble receptor for IL2 (sCD25) ≥2400U/ml.
We present here a series of 5 patients who fulfil clinical criteria of HLH, in which flow cytometric analysis and/or gene sequencing discarded anomalies in perforin as causing the disease. Anomalies in RAB27A, LYST and AP3B1 genes (responsible for Griscelli, Chédiak-Higashi and Hermanski-Pudlak syndromes, respectively) were excluded from the analysis because patients lacked pigmentation defects. Absence of lymphoproliferation excluded BIRC4 gene, which is associated to X-linked lymphoproliferative syndrome.
We aimed at analyzing the genes coding for Munc13-4 and syntaxin 11 proteins in search of mutations that might explain HLH in our patients.
Materials and methodsPatients and sample collectionThe procedures used in this manuscript are in accordance to standards of the Ethical Committee in Hospital 12 de Octubre and with the Helsinki Declaration. Informed consent was obtained from patient's parents at the beginning of the study. All patients were from Spanish genetic background. Most relevant clinical and chemical data are presented in Table 1.
HLH clinical diagnostic criteria in patients 1–5 according to HLH-2004 guidelines (rows 2–9)10 and genetic analysis of patients (rows 10–13).
| HLH clinical criteria10 | PATIENT 1 | PATIENT 2 | PATIENT 3 | PATIENT 4 | PATIENT 5 |
| Fever | Yes | Yes | Yes | Yes | Yes |
| Splenomegaly | Yes | Yes | Yes | Yes | Yes |
| Cytopenias (≥2 peripheral blood lineages) | ThrombocytopeniaLeukopeniaNeutropeniaAnemia | ThrombocytopeniaNeutropeniaAnemia | ThrombocytopeniaAnemia | ThrombocytopeniaLeukopeniaNeutropeniaAnemia | ThrombocytopeniaLeukopeniaNeutropeniaAnemia |
| Hypertrigliceridemia (≥265mg/dl) and/or hypofibrinogenemia (≤1.5g/L) | Tryglicerides335mg/dl | nr | Tryglicerides312mg/dl | Tryglicerides690mg/dl | nr |
| Haemophagocytosis | Yes, in BM | Yes, in BM | nr | Yes, in BM | Yes, in BM |
| Decreased or absent NK activity | Yes | Yes | nr | nr | nr |
| Hyperferritinemia (≥500ng/ml) | 3661ng/ml | nr | 9270ng/ml | 1432ng/ml | nr |
| Soluble IL-2 receptor (sCD25) (≥2400U/ml) | 1383U/ml | 1246U/ml | 5376U/ml | 1687U/ml | nr |
| PRF1 sequencing | Normal | Normal | Normal | Normal | Normal |
| UNC13D sequencing | c.C2782T heterozygous | Normal | IVS9+1G>A splicing mutation in homocigosis | Normal | Normal |
| STX11 sequencing | Normal | Normal | Normal | Normal | Normal |
| STXBP2 sequencing | nt | nt | nt | nt | nt |
nt: not tested; nr: not recorded.
In italics HLH criteria fulfilled in each case.
Expression of intracellular perforin in T and NK cells was tested by using MoAbs anti-CD3-PECy7 (BD Biosciences, San Diego, CA), anti-CD8-PECy5, anti-CD56-PECy5 (Beckman-Coulter, CA, USA) and anti-perforin (δG9 clone) (BD Biosciences), in a Coulter EPICS XL-MCL flow cytometer (Beckman Coulter, CA, USA). Soluble CD25 was tested in serum samples by a commercial ELISA kit (RD, Minneapolis, MN).
NK cytotoxic functionClassical NK cytotoxic assay was performed. Chromium 51 labelled-human K562 cells were cocultured with different ratios of PBMC, and radioactivity was measured with gamma counter. Cytotoxicity was quantified by calculating the percentage specific NK lysis from the formula: cpm (tested)−cpm (spontaneous release)/[cpm (maximum release)−cpm (spontaneous release)]×100.
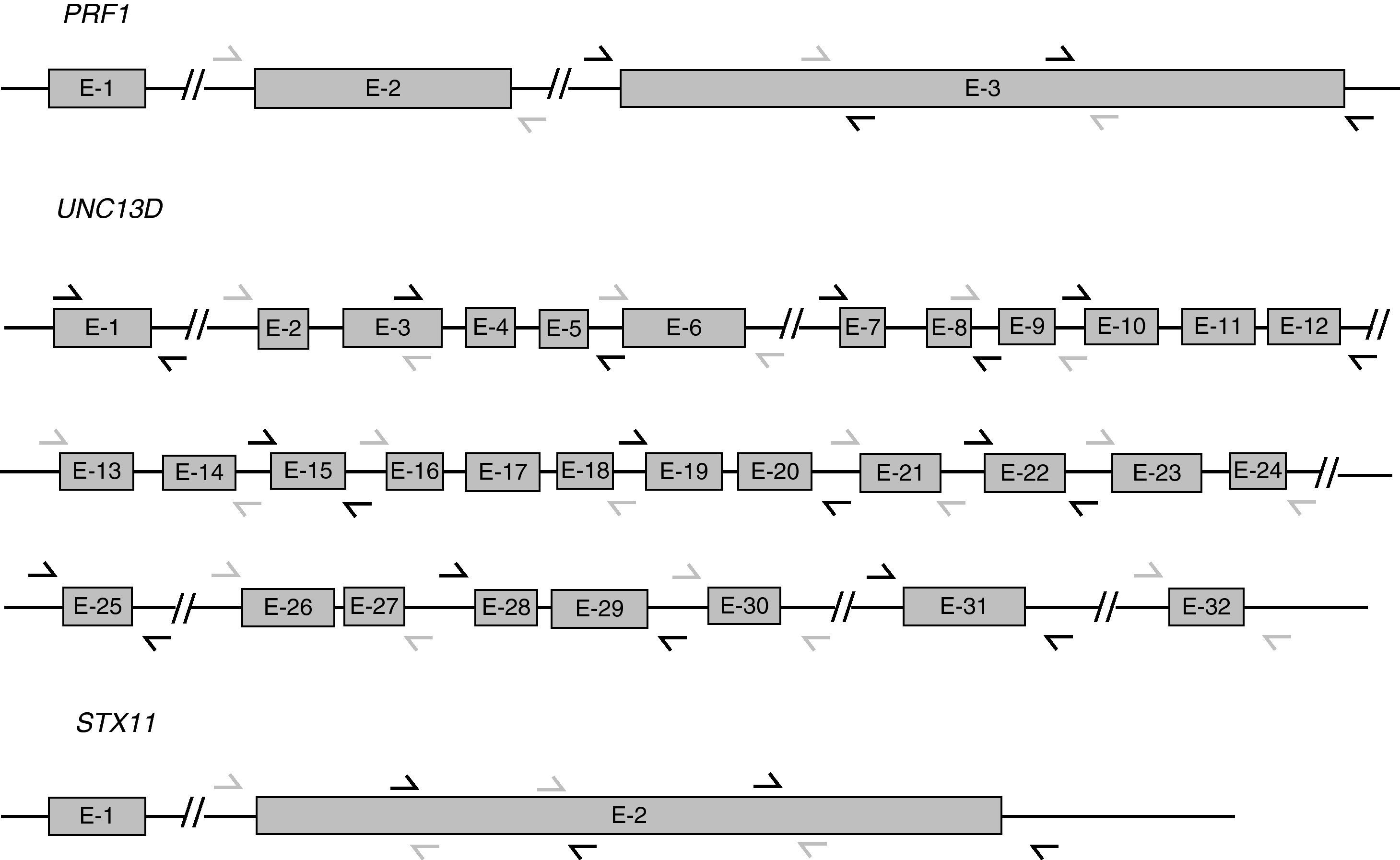
UNC13D and STX11 mutation analysisGenomic DNA was isolated from peripheral blood by standard procedures. For the genetic analysis, the 32 exons of UNC13D, the single coding exon of STX11 (exon 2), and the corresponding surrounding genomic sequences were amplified using polymerase chain reaction (PCR) (Fig. 1). Amplification reactions were performed with 200ng of DNA, 25pmol of each primer, 200μM of dNTPs, 10mM Tris–HCl pH 8.3, 1.5mM MgCl2, 50mM KCl and 1.5U of Taq polymerase, in a final volume of 50μl. Sequences of primers for UNC13D amplification were kindly supplied by Santoro et al.11 Primers for STX11 have been published by Rudd et al.12

The resulting PCR products were purified using the QIAquick PCR purification kit (Qiagen, Hilden, Germany). Purified products were directly sequenced (Sanger's dideoxy chain terminators method, using dye-labelled dideoxy terminators, Applied Biosystem, Foster City, CA). Finally, products were analysed in a DNA sequencer (Applied Biosystem 3100 Avant). STXBP2 gene sequence was not investigated in our study.
ResultsPatient 1A 24-day-old girl was transferred to our center from a regional hospital for investigating hepatosplenomegaly, pancitopenia and cholestasis. She had suffered sepsis and petechial rash at birth. During her hospitalization she presented thrombocytopenia, leukopenia, neutropenia, anemia, repeated episodes of fever (>38°C), erythematous maculopapular rash, hepatosplenomegaly and cholestasis. Abnormal chemistries included hypertrigliceridemia, hyperferritinemia, and high values of sCD25. Bone marrow evaluation revealed some occasional macrophages with haemophagocytosis. In vitro NK cell functional analysis showed decreased NK activity. At month 5 clinical diagnosis of HLH was made based on Histiocyte Society Criteria, and she started on the HLH-94 therapy protocol.10 At month 6 she suffered a severe hemorrhagic shock with hypotension and extreme acidosis that caused an uncontrollable multiorgan failure leading to death.
The expression of intracytoplasmic perforin and DNA sequencing of PRF1 and STX11 genes were normal. Sequencing of UNC13D revealed the heterozygous missense mutation c.C2782T in exon 29 leading to R928C aminoacid change, previously described as a pathogenic mutation.11 The mother was identified as heterozygous carrier. No other mutation was found in patient 1 or in her father.
Because FHL3 has an autosomal recessive inheritance, it can be speculated that a second mutation in UNC13D gene might exist in patient 1. Further genomic DNA or mRNA sequencing could not be performed due to sample limitations. Santoro and co-workers reported that mutations affecting UNC13D mRNA splicing are the most common molecular defects in FHL3 patients.13 The authors identified two deep intronic mutations in IVS1 and in IVS30 and demonstrated that these changes affect regulatory sequences causing aberrant splicing. According to these findings, the patient 1 putative second mutation could be located in a deep intronic regulatory position resulting in a UNC13D splicing defect.
Patient 2A 7-year-old girl was admitted to our hospital by fever of unknown origin. She had been diagnosed of T lymphoblastic leukemia two years before, she had responded well to chemotherapy and was currently in full remission. During hospitalization she presented elevated fever, adenopathy and mild hepatosplenomegaly. Her laboratory data showed anemia and severe thrombocytopenia. Neutrophils count was sometimes below 500 per μl, needing treatment with G-CSF. An infectious origin was discarded. A bone marrow puncture aspiration revealed the presence of numerous macrophages often containing large cellular aggregates and clear phenomena of haemophagocytosis. Additional laboratory data included slightly low NK cells numbers and high levels of sCD25. In vitro NK cell functional analysis showed a decreased NK activity. Flow cytometry analysis of intracytoplasmic perforin expression showed a mild decrease in lymphocytes T CD8+ and NK cells but the sequencing of PRF1 showed no changes compared to the consensus sequence. Testing for mutations in STX11 and UNC13D genes was also negative.
Reduced NK activity could be either secondary to HLH in the context of malignancy and/or immunosuppressive treatment or linked to a genetic defect. Patient 2 case raises the question of whether sporadic cases of HLH harbor a cryptic genetic predisposition. This is shown to be the case in patients with the Ala91Val variant in perforin gene. This variant causes a partial functional impairment of the protein and predisposes to atypical forms of HLH, delayed FHL onset and susceptibility to hematological malignancy.14 A similar mechanism coming from a currently undetected genomic variant could explain the late disease and lymphoma as first FHL manifestation in patient 2.
Patient 3A 42-day-old boy was admitted to the hospital with fever and vomiting. Of note, his family history included a dead sister because of HLH. Several days after admission, he presented hepatosplenomegaly, temperature up to 38.5°C, bicitopenia (thrombocytopenia and anemia), hypertriglyceridemia, and high levels of ferritin. A bone marrow aspirate was made but haemophagocytosis could not be confirmed due to cellular scarcity. Despite lacking a molecular diagnosis of FLH, treatment was initiated according to the international protocol HLH 2004 with good results, achieving a reduction of irritability and hepatosplenomegaly in 48h.
Two years later the patient was readmitted in the hospital due to fever of three days of evolution associated to hepatosplenomegaly, cytopenias and elevated soluble IL-2 receptor, without other laboratory abnormalities. The patient had normal intracytoplasmic perforin expression in CD8+ T lymphocytes and NK cells and normal PRF1 and STX11 sequences. Sequencing of UNC13D genomic DNA revealed a G>A change at position +1 of intron 9. Analysis of cDNA sequence showed that the mutation caused the deletion of exon 9. The patient is currently four years old and remains stable.
Patients 4 and 5Patients 4 and 5 are brothers, presented a family history that included a maternal cousin dead in early childhood with fever, lymphadenopathy and multiorgan failure, and two maternal relatives suffering lymphoma. Patient 4, a 6-year-old girl, was admitted to hospital with clinical suspicion of infectious mononucleosis and positive EBV viremia. Ten days later she presented typical HLH manifestations with severe cytopenias (anemia, thrombocytopenia and leukopenia), fever, hepatosplenomegaly, hypertriglyceridemia, hyperferritinemia, augmented sCD25 and haemophagocytosis in bone marrow. Her deterioration was progressive and she died due to respiratory failure secondary to lung necrosis and hemorrhagic diathesis. Patient 5, a 5-year-old boy, presented also typical HLH manifestations associated with EBV viremia and he died a month and a half after admission due to hemodynamic failure and cardiac arrest. A younger sister of patients 4 and 5 presented infectious mononucleosis at age 4 due to acute EBV infection, but her evolution was good and signs of haemophagocytic syndrome did not developed. Perforin deficiency was discarded at the molecular level in patients and their parents. Sequencing of MUNC13-4 and Syntaxin 11 coding genes in the younger sister and her parents did not reveal any change.
In this family, the remarkable susceptibility to severe EBV disease and its association with HLH prompted differential diagnosis with Duncan disease,15 a primary immunodeficiency characterised by a particular vulnerability to EBV infection leading to malignant lymphoma, dysgammaglobulinemia and frequently to death. This immunodeficiency is caused by mutations in genes SH2D1A/SAP (coding for SLAM-associated protein) and XIAP (coding for X-linked inhibitor or apoptosis) which interfere with important signaling pathways in T lymphocytes, and different HLH forms accompany the disease in 55% and 76% of the cases, respectively.16 Duncan disease diagnosis was discarded in patients 4 and 5 family since patient 4 is a girl and this disease is characteristically linked to X chromosome (XLP, X-linked lymphoproliferative disease).
Recently, Huck and coworkers reported 2 sisters from a consanguineous family who died in childhood after developing severe immune dysregulation and therapy-resistant Epstein-Barr virus positive B-cell proliferation following EBV infection. The authors identified a homozygous mutation in the IL-2-inducible T cell kinase (ITK) gene.17 It may be therefore interesting to study ITK gene in patients 4 and 5, although they do not show lymphoproliferation as a relevant symptom and patients reported by Huck and coworkers did not show HLH.
DiscussionWe present here a series of five patients who fulfil standards for the diagnosis of HLH (Table 1). After analyzing in all five patients the genes more frequently linked to FHL, PRF1, UNC13D, and SXT11, we found a heterozygous missense mutation in UNC13D gene in patient 1 and a homozygous splicing mutation in patient 3. This result agrees with published series in which mutations in UNC13D explained up to 50% of the FLH without PRF1 mutations, suggesting a heterogeneous genetic background for this disease.
That only one mutation in heterozygosis has been found in P1 is probably due to the fact that only exonic regions from genomic DNA were sequenced. It might be speculated that an unidentified mutation is present in the other allele (splicing or other type) and leads to the non-expression of UNC13D. These data have implications in the design of strategies for the genetic analysis of the families with suspected FHL and raise the need to study the UNC13D gene at mRNA level.
A potential limitation of our analysis is the lack of investigation of STXBP2 gene, whose contribution to FHL was unknown at the beginning of the study presented here. STXBP2 gene mutations causing FHL were reported in 20098,9 and were mostly found in Turkish and Middle East populations. More recently, Cetica et al.18 have reported STXBP2 mutations in Caucasian patients from Italy and UK. Investigation of STXBP2 would be especially interesting on P2, and siblings P4 and P5 who did not show mutations in PRF1, UNC13D or STX11. Although we cannot completely discard that these patients presented secondary HLH cases, disease severity, age at onset and family history suggest primary HLH.
Based on published data (some summarized in Table 2), it is estimated that the genetic defect for FHL is not discovered in about 40% of patients. The contribution of the various mutations in FHL-related genes is also different in distinct populations: mutations in PRF1 or UNC13D account for 15% to 50% and 15% to 25% of FHL patients, respectively, depending on geographic region.19 Mutations in STX11 were initially identified in patients of Turkish origin, where they account for approximately 20% of FHL patients. In Turkish population, the genetic defect remains unknown only in 19% of HLH patients.19 In contrast, the molecular base of HLH remains undetected in 70% of German patients.19 The frequency and distribution of the different FHL-related gene anomalies in Spanish population remain presently unknown. Future studies to define additional genetic markers are needed.
FHL patients and families and ethnic background in most relevant publications.
| FHL type and defective gene | % with mutations in the series studied | Ethnic background of series studied | Ethnic background of patients with mutations | Reference |
| FHL1q21.3-q22 region on chromosome 9 | 4 families | Pakistani 100% (total 4 families) | Pakistani 100% (total 4 families) | Ohadi 199923 |
| FHL2PRF1 | 20.6% (7 out of 34 families) | Europe Caucasoids 38%, Turkish 59%, Egypt 3% (total 34 families) | Europe Caucasoids 14%, Turkish 86% (total 7 families) | Göransdotter 2001 |
| 58% (25 out of 43 families) | North American White, Asia, African/American Hispanic (number not specified) | African-American 40% (total 25 families) | Molleran Lee 2004 | |
| 18% (13 out of 76 patients) | Nordic 25%, Middle East 20%, Turkish 55% (total 76 patients) | Nordic 8%, Middle East 46%, Turkish 46% (total 13 patients) | Horne 2008 | |
| FHL3UNC13D | 37.5% (6 out of 16 families negative for PRF1 mutations) | Japanese 100% (total 16 families) | Japanese 37.5% (total 6 families) | Yamamoto 2004 |
| 50% (15 out of 30 families negative for PRF1 mutations) | Italian 87%, Others 13% (total 30 families) | Italian 80%, Others 20% (total 15 families) | Santoro 2006 | |
| 28.6% (18 out of 63 patients) | Turkish 51%, German 36%, Others 13% (total 63 patients) | Turkish 44%, German 33%, Others 23% (total 18 patients) | Zur Stadt 2006 | |
| FHL4STX11 | 9.5% (6 out of 63 patients) | Turkish 51%, German 36%, Others 13% (total 63 patients) | Turkish 100% (total 6 patients) | Zur Stadt 2006 |
| 14% (4 out of 28 families negative for PRF1 mutations) | Turkish 68%, Europe+Arabian 32% (total 28 families) | Turkish 100% (total 4 patients) | Rudd 2006 | |
| FHL5UNC18-2 | 100% (6 out of 6 families without PRF1, UNC13D, STX11 mutation) | Saudi Arabian 50%, Palestinian 16.7%, Turkish 16.7%, Iranian 10.7% (total 6 families) | Saudi Arabian 50%, Palestinian 16.7%, Turkish 16.7%, Iranian 16.7% (total 6 families) | Côte 2009 |
| 60% (9 out of 15 patients without PRF1, UNC13D, STX11 mutations) | Turkish+Saudi Arabian 100% (total 15 patients) | Turkish 67%, Arabian 33% (total 9 patients) | Zur Stadt 2009 | |
| 14% (4 out of 28 families without PRF1, UNC13D or STX 11 mutations) | Italian 61%, UK 14%, Others 25% (total 28 families) | 25% Italian, 25% Pakistani, 25% Kuwaiti, 25% UK (total 4 patients) | Cetica 2010 | |
Because sequencing of the genes, especially UNC13D is resources- and time-consuming, flow cytometric analysis, Western blot and functional assays should be used as previous screening tools to guide the genetic study. NK cell function is one of the most useful laboratory tests for HLH diagnosis, as it is diminished in up to 90% of patients. However, because a reduced NK activity can be found in primary as well as in secondary HLH, NK function test does not distinguish between both HLH forms. Flow cytometry is useful to detect perforin defects. The degranulation capacity of lymphocytes can be measured by expression of CD107a, an integral lysosomal membrane protein that is expressed on the surface of cytotoxic lymphocytes upon exocytosis. The degranulation assay is useful for the detection of patients with UNC13D, STX11 and STXBP2 mutations.8,9,20–22 Finally, extensive genetic analysis is mandatory for a correct diagnosis and for the indication for haematopoietic stem cell transplantation and selection of familial donor, or for the consideration of alternative, currently emerging therapeutical approaches specifically targeting activation of NK or CD8+ T cells.21
Grant sponsorThis work was supported by grants from Fondo de Investigación Sanitaria FIS PI06/0170 and PI06/0614, Ministerio de Sanidad y Consumo, Spain.
Conflict of interestsAuthors declare no conflict of interests in the manuscript. The financing sources have not participated in the design, data collection, analysis or interpretation of data, in the manuscript writing or in the decision to submit for publication.