





La deficiencia selectiva de Inmunoglobulina A (IgA del suero < 0.07g/l) es la inmunodeficiencia primaria más frecuente, generalmente asintomática. Se conoce muy poco acerca de los mecanismos etiopatogénicos que conducen a esta anormalidad inmunológica. La deficiencia selectiva de Inmunoglobulina A (DIgA) está ligada a una enfermedad de producción de inmunoglobulina más severa: inmunodeficiencia común variable (IDCV). La ocurrencia familiar de ambas enfermedades ha sido documentada repetidamente. Se presentan 5 pacientes femeninas de 10, 16, 20, 30 y 47 años de edad con antecedentes patológicos personales y familiares de atopia, valoradas por consulta de inmunología por infecciones recurrentes que no responden al tratamiento antibiótico habitual, púrpura vascular, dolor articular y cianosis distal en la paciente de 47 años de edad. Los estudios inmunológicos revelaron IgA ausente en las 5 pacientes, y en la de 47 años de edad una disminución de las concentraciones séricas de IgG, y un incremento de los números relativos de células CD3+, CD4+, CD8+ en relación a los otros 4 casos.
Selective deficiency of immunoglobulin A (serum IgA < 0.07g/l) is the most frequent primary immunodeficiency, usually asymptomatic. Very little is known about the aetiopathogenic mechanism leading to this immunological abnormality. Selective deficiency of immunoglobulin A (IgAD) is linked to a more severe immunoglobulin production disease: common variable immunodeficiency (CVID). Familial occurrence of both diseases has been repeatedly documented. We present five cases of 10, 16, 20, 30 and 47 years-old females with personal and familial pathologic antecedents of atopy, evaluated by immunology for recurrent infection that do no resolve with common antibiotic treatment, as well as vascular purple, articular pain and distal cyanosis in the 47 years-old case. The immunologic studies reveals absent IgA in the five cases and, in the 47 yearsold case a lower serum concentration of IgG and increased relative numbers of CD3+, CD4+, CD8+ cells in relation to the other four cases.
La deficiencia selectiva de IgA (IgA del suero< 0,07 g/l) es la inmunodeficiencia primaria más frecuente pero usual-mente asintomática y se caracteriza por concentraciones en el suero de IgA disminuidas y niveles normales de IgG e IgM(1,2), aunque algunos pacientes son afectados por infecciones repetidas del tracto respiratorio superior y/o gastrointestinal(3). A pesar de su alta frecuencia, se conoce muy poco acerca de los mecanismos etiopatogénicos que conducen a esta anormalidad inmunológica(4).
Los individuos deficientes de IgA pueden ser inmunizados y desarrollar anticuerpos anti-IgA, y en las transfusiones subsiguientes de productos de la sangre que contienen proteína IgA pueden producirse reacciones anafilácticas que ponen en peligro la vida. Los anticuerpos contra IgA están presentes en el 44% de individuos deficientes de IgA(5).
La prevalencia de la deficiencia de IgA selectiva varía desde 1:320 en EE.UU.(6) a 1:18, 500 en Japón(7), dependiendo del trasfondo étnico, sugiriendo una base genética para el trastorno. Alrededor de un individuo en 600 carece de IgA en el suero y en las secreciones pero tiene niveles normales de los otros isotipos de inmunoglobulinas(8). Los individuos deficientes de IgA son usualmente asintomáticos, y por tanto son raramente diagnosticados; sus deficiencias de IgA son descubiertas accidentalmente, por ejemplo: en pesquisaje de donantes de sangre. Es importante conocer su prevalencia, ya que ayuda a los bancos de sangre a asegurar la necesidad de tener un pool de donantes deficientes de IgA(9,10).
La deficiencia selectiva de inmunoglobulina A (DIgA) está ligada a una enfermedad de producción de inmunoglobulina más severa: la inmunodeficiencia común variable (IDCV), que se caracteriza por niveles de IgG e IgA disminuidos mientras los niveles de IgM son variables, y números de células B normales o disminuidos conduciendo a infecciones recurrentes. La inmunodeficiencia clínicamente significativa aparece a cualquier edad después de una función aparente normal del sistema inmune(11,12). La ocurrencia familiar de ambas enfermedades ha sido documentada repetidamente(12,13). El patrón hereditario de ambas enfermedades es variable, en algunas familias se ha demostrado herencia autosómica recesiva o dominante, siendo más comunes los casos esporádicos y su asociación a algunos haplotipos HLA determinados por el complejo principal de histocompatibilidad (MHC)(14-17). Al menos dos loci distintos, uno en la región clase II y uno en la región clase III del complejo MHC confieren susceptibilidad al desarrollo de DIgA e IDCV(18). Se ha demostrado que los haplo-tipos HLA-A1, B8, DR3 extendidos están asociado con DIgA e IDCV(19). Afectan en igual proporción a hombres y mujeres.
La inmunodeficiencia común variable (IDCV) es la inmunodeficiencia primaria con hipogammaglobulinemia que más frecuentemente ocurre; otros ejemplos de inmunodeficiencias primarias con hipogammaglobulinemia son la hipogammaglobulinemia transitoria de la infancia, la agammaglobulinemia ligada al cromosoma X (ALX), inmunodeficiencia combinada severa (IDCS), síndrome de hiper IgM ligado-X, y varios tipos de defectos de genes autosómicos recesivos con trastornos de la maduración de células B(20).
En este trabajo se presentan 4 casos de déficit selectivo de IgA y uno de inmunodeficiencia común variable, inmunodeficiencias primarias de inmunoglobulinas.
MATERIALES Y MÉTODOSPreviamente a la realización del reporte de los 5 casos se obtuvo el consentimiento por escrito de las pacientes y de la madre de la paciente de 10 años de edad, quienes estuvieron de acuerdo con la realización de estudios inmunológicos con toma de muestras de sangre, estudios de ultrasonido abdominal y la obtención de fotos de región cutánea de las pacientes afectadas en el momento del estudio. Se incluyeron 4 pacientes con el diagnóstico de Déficit Selectivo de IgA y una paciente de 47 años de edad con el diagnóstico probable de Inmunodeficiencia Común Variable.
Todas las determinaciones fueron realizadas a ciegas en el laboratorio clínico, como indicación habitual a los pacientes que acuden a consulta de inmunología con relación a la clínica del paciente.
Se determinó la cuantificación de inmunoglobulinas(21), Proteína C reactiva, C3 y C4, factor reumatoideo (Roche Diagnostics GmbH, Mannheim Germany/Hitachi 902), inmunocomplejos circulantes(22), ANA(23). Se estudió la inmu-nidad celular mediante la determinación de subpoblacio-nes linfocitarias por citometría de flujo(24).
CASOS CLÍNICOSCaso 1Paciente femenina de 10 años de edad, con antecedentes patológicos personales de rinitis alérgica y antecedentes familiares de atopia (tíos paternos asma bronquial persistente moderada). Al año de edad comenzó con infecciones respiratorias altas a repetición (faringoamigdalitis, gripes) de evolución prolongada. A los 7 años presentó un eritema fijo medicamentoso.
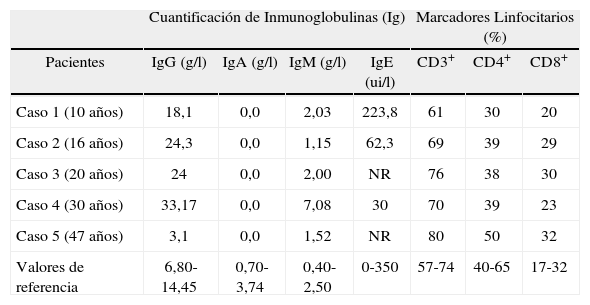
Los estudios inmunológicos revelaron ausencia de IgA, IgG elevada e IgM normal (IgG: 18,1 g/l, IgA: 0,0 g/l e IgM: 2,03 g/l); niveles normales de IgE (223,8 ui/ml), subpoblaciones linfocitarias CD3+: 61%, CD4+: 30%, CD8+: 20% (Tabla I), C3 y C4 normal (C3: 1,65 g/l y C4: 0,23 g/l).
Niveles de inmunoglobulinas y subpoblaciones linfocitarias
| Cuantificación de Inmunoglobulinas (Ig) | Marcadores Linfocitarios (%) | ||||||
| Pacientes | IgG (g/l) | IgA (g/l) | IgM (g/l) | IgE (ui/l) | CD3+ | CD4+ | CD8+ |
| Caso 1 (10 años) | 18,1 | 0,0 | 2,03 | 223,8 | 61 | 30 | 20 |
| Caso 2 (16 años) | 24,3 | 0,0 | 1,15 | 62,3 | 69 | 39 | 29 |
| Caso 3 (20 años) | 24 | 0,0 | 2,00 | NR | 76 | 38 | 30 |
| Caso 4 (30 años) | 33,17 | 0,0 | 7,08 | 30 | 70 | 39 | 23 |
| Caso 5 (47 años) | 3,1 | 0,0 | 1,52 | NR | 80 | 50 | 32 |
| Valores de referencia | 6,80-14,45 | 0,70-3,74 | 0,40-2,50 | 0-350 | 57-74 | 40-65 | 17-32 |
NR: No realizado.
Paciente femenina de 16 años de edad, con antecedentes patológicos personales de rinitis alérgica, dermatitis a los 15 años de edad, huella del BCG, hipotonía muscular al nacer, escoliosis, pie plano y valgo, artralgia en miembros inferiores y con antecedentes familiares de atopia (padre rinitis y bronquitis, hermano coriza y bronquitis) y hermano con glaucoma. En la niñez temprana presentó gripes frecuentes, hospitalización por neumonías en dos ocasiones. A los dos años se operó de adenoides y comenzó a presentar faringoamigdalitis a repetición, tratándose con penicilina benzatínica con mejoría del cuadro clínico. Actualmente ha presentado faringoamigdalitis y cuadros gripales mensuales. Desde la niñez las enfermedades infecciosas demoran más en curarse que en otras personas de su grupo etario y siempre hay que usar antibiótico para resolver el cuadro, registrado en historia clínica de la paciente. Presenta dolores articulares en miembros inferiores (principalmente en las piernas). Además, en estos momentos presenta lesiones nodulares eritematosas en ambas axilas, con aumento de la temperatura local, dolorosas, una de ellas con pústula central (hidrosadenitis) y forúnculos en el antebrazo y en la pierna, tratados con antibiótico, que drenaron espontáneamente durante el tratamiento. Los estudios de laboratorio mostraron Proteína C reactiva: 10,6 mg/l (ligeramente elevada), Hb: 13,2 g/l, leucocitos: 9,7 x 109/l, plaquetas: 3 79 x 109/l, Linfocitos: 41,9%, Monocitos 4,3%, polimorfonucleares 53,8%. Los estudios inmunológicos revelaron ausencia de IgA, IgG elevada e IgM normal (IgG: 24,3 g/l, IgA: 0,0 g/l e IgM: 1,15 g/l); niveles normales de IgE (62,3 ui/ml); subpoblaciones linfocitarias CD3+: 69%; CD4+: 39%; CD8+: 29% (Tabla I); FR: negativo; C3 y C4 normal (C3: 1,42 g/l y C4: 0,11 g/l). El ultrasonido de hemiabdomen superior mostró hígado de tamaño y textura normal; Bazo normal.
Caso 3Paciente femenina de 20 años de edad, con antecedentes patológicos personales de asma bronquial persistente severa, anomalía congénita (costillas supernumerarias), y con antecedentes familiares de atopia, madre con fibromialgia reumática y costillas supernumerarias, y padre con atrofia renal derecha. A los 2 años presentó una bronco-neumonía en ambos campos pulmonares con ingreso en unidad de cuidados intensivos (UCI). Además presentó infecciones respiratorias altas a repetición (faringoamigdalitis frecuentes lo que conllevo tratamiento quirúrgico: amigdalectomía), otitis derecha. A los 14 años presentó una varicela complicada, que duró 15 días con pústulas generalizadas en piel y mucosas. A los 15 años presentó abscesos en diferentes regiones del cuerpo: inguinal, párpado inferior del ojo izquierdo, en el dedo anular de la mano izquierda y dos abscesos en el muslo izquierdo, que no respondieron al tratamiento antibiótico habitual. Además, sufrió dengue clásico a los 18 años y actualmente amigdalitis aisladas en tejido remanente de amígdala. En la actualidad, la paciente presenta crisis de diarreas crónicas, sin etiología precisada. Los estudios inmunológicos revelaron ausencia de IgA, IgG elevada e IgM normal (IgG: 24,0 g/l, IgA: 0,0 g/l e IgM: 2,0 g/l); subpoblaciones linfocitarias CD3+: 76%, CD4+: 38%, CD8+: 30% (Tabla I).
Caso 4Paciente femenina de 30 años de edad con antecedentes patológicos personales de coriza, hepatitis en la infancia; con antecedentes familiares de atopia, madre con asma bronquial persistente severa, hepatitis B complicada y padre asmático persistente leve. Durante el segundo embarazo sufrió vaginosis bacteriana y después del mismo presenta episodios de foliculitis que no responden al tratamiento antibiótico, además de micosis ungueal frecuente en los pies. Actualmente presenta lesiones papulopustulosas y eritematosas con intensa reacción celulítica alrededor de las mismas, con pústulas grandes de color amarillento, dolorosas, que al involucionar dejan lesiones hiperpigmentadas y cicatrizales deprimidas en abdomen y pierna izquierda (Fig. 1). Los estudios de laboratorio al comienzo de las infecciones mostraron hemoglobina 12,4 g/l, leucocitos 8,6 x 109/l, polimorfonucleares 49 %, linfocitos 45 %, eosinófilos 3 %. La eritrosedimentación 44 mm/1h. Los estudios inmunológicos revelaron ausencia de IgA, con IgG e IgM elevadas (IgG: 33,17 g/l, IgA: 0,0 g/l, IgM: 7,08 g/l), niveles normales de IgE (30 ui/ml); subpoblaciones linfocitarias CD3+: 70%, CD4+: 39%, CD8+: 23% (Tabla I), proteína C reactiva negativa. En el exudado cutáneo se detecta Staphylococcus aureus. Se realiza biopsia cutánea, con diagnóstico anatomopatológico: Fragmento de tejido cutáneo que muestra infiltrado inflamatorio agudo perianexial que se extiende hasta el tejido celular subcutáneo (TCS), con destrucción de un folículo piloso. El aspecto histológico es compatible con una foliculitis aguda.
Caso 5 Pierna. B) Muslo.")
Paciente femenina de 47 años de edad con antecedentes patológicos personales de otitis y sinusitis maxilar en la infancia, asma bronquial persistente moderada, rinitis alérgica que aparece a los 23 años. Presenta además intolerancia a la aspirina, a AINE (antiinflamatorios no esteroideos) y eritema fijo medicamentoso a sulfamidas, metronidazol, tetraciclina, penicilina y dipirona. No hay huella del BCG, sufre un aborto espontáneo a los 30 años de edad, hipertensión arterial durante sus dos embarazos, y antecedentes familiares de atopia. La madre sufre Lupus eritematoso sistémico (LES) y Tiroiditis de Hashimoto (operada de Tiroides). Padre fallecido por una úlcera complicada, tío materno asmático persistente leve y diabético, hermano asmático persistente moderado. Desde hace 2 años presenta episodios de sepsis oral, faringoamigdalitis, giardiasis, diarreas, dolores articulares y de columna que se ha valorado osteopenia por densitometría. Además sufre micosis inguinal resistente al tratamiento habitual, y herpes a repetición. Actualmente, debuta con lesiones purpúreas en miembros inferiores (de las rodillas a los tobillos) acompañadas de dolor abdominal a tipo cólico, sin relación causal medicamentosa, con diagnóstico de púrpura vascular; que cedió con esteroides. Además presenta episodios de cianosis y dolor de dedos y manos con la exposición al frio. En piel presenta lesiones eritematoescamosas con bordes precisos y pruriginosas en axilas, cuero cabelludo (región posterior del cuello), palmas de las manos, caderas y muslos, de 1 mes de evolución (Fig. 2). Se realiza biopsia cutánea, con diagnóstico anatomopatológico de dermatitis psoriasiforme superficial con presencia de microvesículas intraepidérmicas y algunos queratinocitos apoptoticos. Infiltrado inflamatorio linfocitario perivascular e intersticial en el dermis superior. El aspecto histopatológico es compatible con una dermatitis subaguda. Las técnicas de PAS y Plata para hongos fueron negativas. Los estudios de laboratorio al comienzo del cuadro clínico mostraron: Inmunocomplejos circulantes: 0,051; ANA: Negativo; Proteína C reactiva: 1,4 mg/l, Hb: 12,3 g/l; Eritrosedimentación: 20 mm/h, Leucocitos: 3,8x109/l (leucopenia); plaquetas: 267x109/l; Linfocitos: 42,5%; Monocitos: 6%; polimorfonucleares neutrófilos: 51,5%. Los estu dios inmunológicos revelaron una disminución de las concentraciones séricas de IgG, IgA ausente e IgM normal (IgG: 3,1 g/l, IgA: 0,0 g/l e IgM: 1,52 g/l); subpoblaciones linfocitarias CD3+: 80%, CD4+: 50%, CD8+: 32% (Tabla I); Factor Reumatoideo (FR): negativo. El ultrasonido de hemiabdomen superior mostró hígado de tamaño y textura normal y bazo normal.
DISCUSIÓN Muslo. B) Mano.")
El déficit selectivo de IgA y la inmunodeficiencia común variable se definen entre las formas más comunes de inmunodeficiencia primaria.
La ocurrencia del déficit selectivo de IgA y la IDCV ha sido reportada en familias, existe un fuerte agrupamiento natural de ambas enfermedades y la similitud de los defectos subsiguientes de células B, lo que sugiere el involucramiento de factores genéticos no identificados en la patogénesis de ambas. La deficiencia de activador transmembrana e interactor CALM (TACI) se observó en pacientes con IDCV y DIgA(25,26). Todos estos resultados muestran que la DIgA y la IDCV pueden ser manifestaciones fenotípicas diferentes del mismo, o similar, fondo genético. La progresión de DIgA en IDCV reportada en varios casos, muestra que la DIgA puede ser un estadio de desarrollo en la evolución a IDCV(27). Los casos estudiados aquí tienen ascen dencia española (europea), excepto un caso que se desconoce (Tabla II).
Comparación de las alteraciones de los cinco casos
| Caso 1 | Caso 2 | Caso 3 | Caso 4 | Caso 5 | |
| Edad | 10 años | 16 años | 20 años | 30 años | 47 años |
| Raza | Mestiza | Blanca | Blanca | Blanca | Blanca |
| Ascendencia | Española (Tatarabuelos maternos y paternos) | Española (Bisabuelo materno) | Española (Bisabuelos maternos y paternos) | No se conoce | Española (Bisabuelos maternos) |
| APF (Atopia) | Sí | Sí | Sí | Sí | Sí |
| APF (Autoinmune) | No | No | Sí | No | Sí |
| Tipo de alergia | Rinitis alérgica | Rinitis alérgica | Asma bronquial | Coriza | Asma bronquial, Rinitis alérgica |
| Huella BCG | Sí | Sí | Sí | Sí | No |
| Infecciones | Sí | Sí | Sí | Sí | Sí |
| Respiratorias altas | Faringoamigdalitis, Gripes | Faringoamigdalitis, Gripes | Faringoamigdalitis | No | Faringoamigdalitis |
| Senopulmonares | No | Neumonias | Bronconeumonia | No | Sinusitis maxilar (niña) |
| Oídos | No | No | Otitis | No | Otitis (niña) |
| Tracto intestinal | No | Giardiasis, Diarreas | Giardiasis, Diarreas | No | Giardiasis, Diarreas |
| Abscesos | No | Sí | Sí | Sí | No |
| Otras | No | Forúnculos, Hidrosadenitis | Varicela complicada, Dengue | Vaginosis bacteriana, Micosis, Foliculitis | Sepsis oral, Herpes a repetición |
| Piel | Eritema fijo medicamentoso | Dermatitis atópica | Dermatitis psoriasiforme | No | Dermatitis psoriasiforme, Eritema fijo medicamentoso, Púrpura vascular |
| Artralgia | No | Sí | No | No | Sí |
| Anomalías congénitas | No | No | Costillas supernumerarias | No | No |
| Cianosis distal | No | No | No | No | Sí |
| Inmunodeficiencia | Sí | Sí | Sí | Sí | Sí |
| IgG | Elevada | Elevada | Elevada | Elevada | Baja |
| IgA | Ausente | Ausente | Ausente | Ausente | Ausente |
| IgM | Normal | Normal | Normal | Elevada | Normal |
Determinados pacientes con Déficit selectivo de IgA (DIgA) muestran una frecuencia incrementada de infecciones, alergias y manifestaciones autoinmunes(28,29); el defecto parece ser el resultado de un deterioro en el cambio a IgA o un fallo en la maduración de los linfocitos que producen IgA. Los 5 casos estudiados tienen en común ausencia de IgA en el suero, antecedentes patológicos personales y familiares de atopia (el caso 3 y 5 presentan asma bronquial persistente severa y moderada), e infecciones a diferentes niveles: infecciones respiratorias (faringoamigdalitis, cuadros gripales, otitis, infecciones senopulmonares), infecciones cutáneas (forúnculos, foliculitis, abscesos, hidrosadenitis, varicela complicada, y micosis ungueal), y a nivel de mucosa oral y vaginal (vaginosis bacteriana, sepsis oral y herpes virus), además de infecciones en el aparato gastrointestinal por protozoos (giardiasis). Se observa en los cinco casos un predominio de las infecciones piógenas, en los casos 2, 3 y 5 infecciones virales y parasitarias, y micóticas en el 4. Otras manifestaciones cutáneas no infecciosas fueron dermatitis atópica en el caso 2, dermatitis psoriasiforme en los casos 3 y 5, eritema fijo medicamentoso, púrpura vascular y cianosis distal en el caso 5, la artralgia de expresión clínica en los casos 2 y 5, y anomalía congénita en el caso 3 (Tabla II). Por lo que se puede apreciar los cinco casos presentan infecciones y atopia, existiendo una gran similitud entre los casos 2, 3 y 5, por lo que los casos 2 y 3 con DIgA pudieran estar en un estadio del desarrollo en la evolución a IDCV.
La inmunodeficiencia común variable (IDCV) es una enfermedad primaria de deficiencia de anticuerpos que muestra muchas características clínicas similares con la Deficiencia de IgA (DIgA). También, se ha reportado en varios casos la progresión del DIgA a IDCV. A Aghamohammadi y colaboradores presentaron 4 casos con DIgA y características autoinmunes quienes posteriormente desarrollaron IDCV(29). Todos los pacientes con DIgA sintomáticos, especialmente aquellos con deficiencia de subclases de IgG asociadas o características autoinmunes, deben ser monitoreados para evolución a IDCV. La co-ocurrencia de psoriasis y miastenia gravis en cuatro pacientes y la frecuencia de 42% de pacientes con déficit de IgA con desórdenes autoinmunes sugieren que la autoinmunidad podría ser un factor de riesgo para progresión a IDCV. El diagnóstico temprano de esta conversión y la institución de terapia de inmunoglobulina son efectivas en la prevención de infecciones bacterianas severas e insuficiencia pulmonar(27,29).
Las concentraciones séricas de Inmunoglobulina G estaban incrementadas en los casos 1 al 4 y eran bajas en el 5, y las de IgM normales en los casos 1, 2, 3 y 5, e incrementada en el 4. La IgA estaba ausente en los cinco casos, como se describió anteriormente.
Estudios previos realizados en un número limitado de pacientes mostraron un posible decrecimiento de la proporción de células CD4+ y un incremento de la proporción de células CD8+(30-32), mientras que Klemola y colaboradores(33) no observaron anormalidades significativas en los números relativos de células CD3+, CD4+ y CD8+. Debido al similar fondo genético mencionado anteriormente de la IDCV y el defecto de IgA, explicar aquellas anormalidades en el DIgA pudieran ayudar a dilucidar si las anormalidades linfocíticas en la IDCV son primarias, heredadas o si evolucionan secundariamente durante el desarrollo de las enfermedades, o son una consecuencia de la inmunodeficiencia en esta enfermedad.
El estudio de Litzman y colaboradores mostró que los números relativos de células CD3+, CD8+ estaban incrementados en los pacientes con IDCV mientras las células CD4+ estaban disminuidas en relación a los pacientes con déficit selectivo de IgA(34). En los casos estudiados aquí los números relativos de células CD3+, CD4+, CD8+ estaban incrementados en el caso 5 en relación a los otros 4 casos (Tabla I).
Una plétora de anormalidades en las supoblaciones linfocitarias y la expresión de marcadores de activación fueron documentadas reiteradamente en pacientes con IDCV mientras que los datos disponibles acerca de las subpoblaciones linfocitarias en los pacientes con DIgA son escasos. En un estudio realizado por L Hummelshoj y colaboradores se determinaron las subpoblaciones básicas de linfocitos, y aquellas subpoblaciones que se han reportado como anormales en pacientes con IDCV (CD25, antígeno leucocitario humano (HLA)-DR, CD45RA, CD45RO, CD27, CD28 y CD29 en ambas células CD4+ y CD8+, CD57 y CD38 en células CD8+, CD21, CD27, IgM, IgD en linfocitos B) en 85 pacientes con DIgA, 47 pacientes con IDCV y en 65 controles sanos. Los resultados de ese estudio mostraron un incremento en el número relativo de células CD8+ y una disminución en el número absoluto de células CD4+ comparado a la población sana, pero anormalidades similares en pacientes con IDCV fueron mucho más expresadas. Los pacientes con DIgA tenían significativamente disminuida la expresión de HLA-DR y la expresión incrementada de CD25 en los linfocitos CD4+, también la expresión de CD29 estaba disminuida en las células CD8+, mientras no hubo cambios en la expresión de otros marcadores de activación/diferenciación en las células T (incluyendo la expresión de los antígenos CD45RA y CD45RO). No hubo anormalidades estadísticamente significativas en los estadios de desarrollo de linfocitos B en pacientes con DIgA comparados con los controles sanos. La observación mostró que la mayoría de las anormalidades de las subpoblaciones de linfocitos B y T descritas anteriormente en IDCV no están presentes en pacientes DIgA(34,35).
El monitoreo de estos pacientes podría ser efectivo para la limitación de los signos y síntomas; por lo que podría recomendarse que todos los pacientes con Déficit IgA sintomáticos deberían monitorearse para posible progresión a IDCV.
AGRADECIMIENTOSLos autores agradecen a la Dra. Teresa Español por su oportuna crítica y a las pacientes por su cooperación. A la licenciada en Bibliotecología Rafaela Santana y Mirtha Gómez, de la Biblioteca del Hospital por su apoyo, y al informático Jorge Batista por las fotografías.
CONFLICTO DE INTERESLos autores declaran no tener conflicto de interés.