


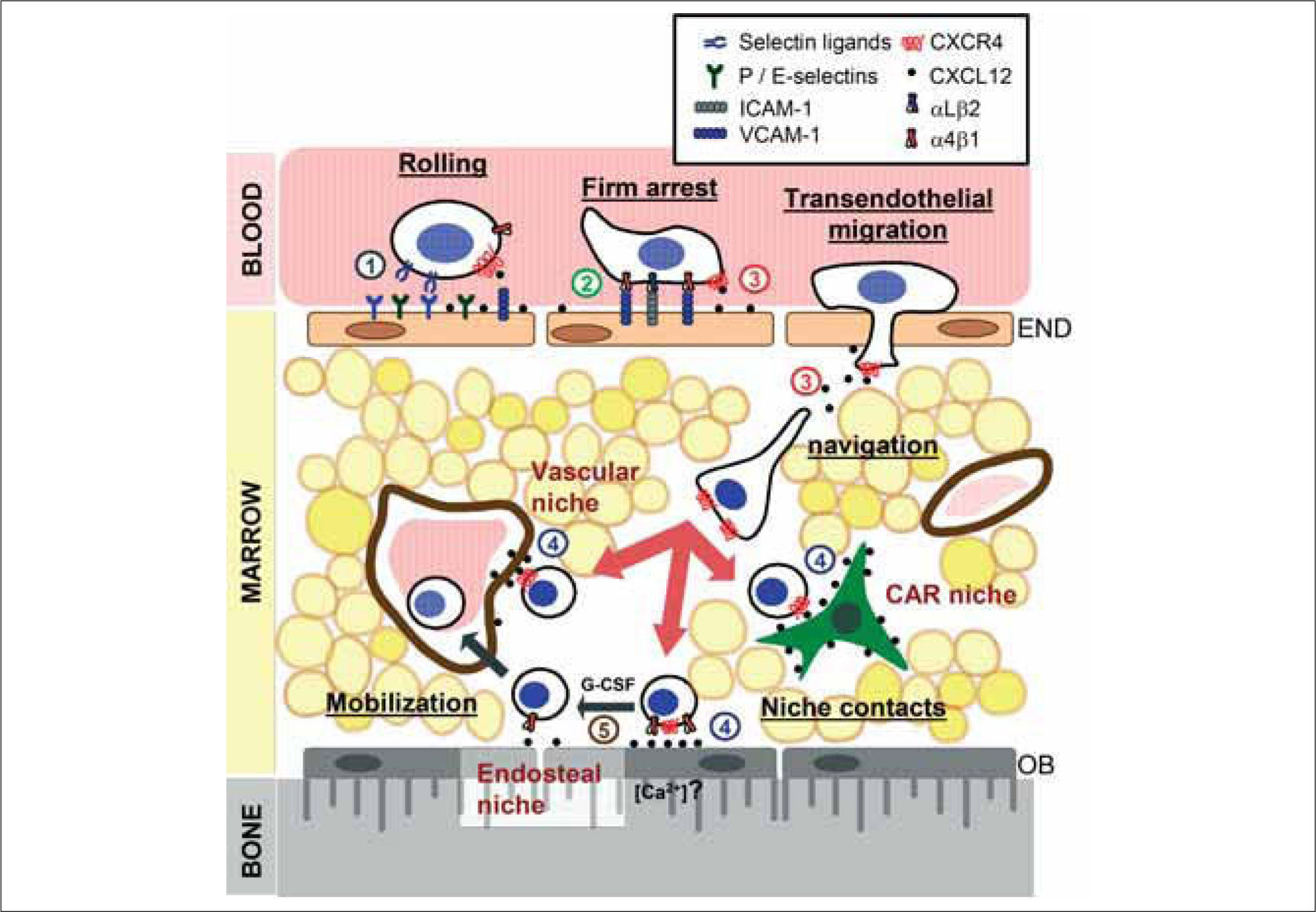
In adult mammals, a rare population of cells that gives rise to the hematopoietic compartment dwells in niches within the bone marrow where they are nourished and protected. These blood stem cells can, however, exit these niches under physiological conditions and, in the context of bone marrow transplantation, migrate to the recipient's marrow to regenerate hematopoiesis. This review will address old and new discoveries regarding the mechanisms that allow these stem cells to find their way home through long and winding roads: From the peripheral circulation, through interactions with the vessel-lining endothelial cells in the bone marrow, and into their niches located in the intramedullary space. It will also address how these discoveries have provided us with new tools to manipulate stem cell trafficking.
En mamíferos adultos, una población celular muy restringida que da origen al tejido hematopoyético se localiza en nichos dentro de la médula ósea en los que recibe sustento y protección. Estas células madre hematopoiéticas de la sangre pueden salir de estos nichos en condiciones fisiológicas y, en el contexto del transplante de médula ósea, migrar a la médula del recipiente para regenerar su sistema hematopoyético. En esta revisión se describen las evidencias que han permitido caracterizar los mecanismos por los cuales estas células madre migran a los nichos medulares siguiendo un camino largo y tortuoso: Desde la circulación periférica, y mediante interacciones con la vasculatura medular, hasta nichos localizados en el espacio intramedular. También se discute cómo se han generado nuevas herramientas que nos permitirán manipular el tráfico de células madre.
Hematopoietic stem and progenitor cells (HSPC) are determined travelers. Beginning from their embryonic origins, HSPC move from one niche to another: The first wave of adult-type hematopoietic progenitors in the mouse appears in the aorta-gonad-mesonephros region around embryonic day 9 (E9), and by E10.5 they relocate to the fetal liver and then to the spleen and the bone marrow (BM)(1). After birth, the BM provides the definitive lodgment for HSPC. The choice of this residence is not casual: The surrounding bone provides physical protection from external aggressions, and cellular elements of the marrow nourish and maintain HSPC. The latter constitute the so-called "stem cell niche", whose components we are only now starting to identify(2). Because this niche has the basic features of what we would define as a home, the migration of HSPC to the BM is commonly referred to as homing.
Three different locations and components of this niche have been reported. Osteoblasts lining the endosteal surface of the bone were the first cellular niche to be identified though elegant genetic approaches in the mouse(3,4). Novel tools that specifically label HSPC later provided visual demonstration of their localization in close contact with endothelial cells lining the vasculature of the BM and spleen(5). More recently, a reticular-like cell type scattered in the medullary parenchyma to which HSPC associated was identified(6). These cells were identified on the basis of being the main producers of CXCL12 (also known as stromal-cell derived factor-1, or SDF-1), a chemokine essential for HSPC retention in the BM. They were thus named CXCL12-abundant reticular (CAR) cells(6). Although this review does not intend to cover details about the HSPC niche, it is important to understand the basic features of the destination of the road that HSPC follow which such remarkable determination.
Because the niche is so important for the survival and proper differentiation of HSPC, one might ask why these cells would ever leave such a protective environment. Is there a need for HSPC to migrate or home back to this niche? Why is this process worth studying?
It has been known for over 40 years that HSPC are present in the circulating blood under physiological conditions(7,8). Although dependent on several factors, such as strain or pathophysiological state, the number of HSPC in the blood of a mouse at any given moment ranges from 10 to 60 (Daniel Lucas-Alcaraz and Paul Frenette, personal communication). These circulating HSPC are functional in that they are capable of restoring hematopoiesis as demonstrated in experiments using competitive reconstituting assays and parabiotic mice(9,10). Although the significance of these observations is presently unclear, these experiments demonstrated for the first time that HSPC routinely exit and re-enter the BM.
The relevance of this process is perhaps most salient in the clinical arena. The ability of HSPC to specifically home to the BM was first demonstrated in the early 1950s in experiments showing that pluripotent cells present in the spleen or BM were capable of repopulating hematopoietic organs(11,12). These observations paved the way toward the clinical use of bone marrow transplantation (BMT) to treat hematological malignancies(13). BMT is currently a common procedure used to restore hematopoiesis in patients undergoing myeloablation, and has been also used for gene therapy in patients suffering anemia and immunodeficiencies(14,15). In this context, HSPC obtained from a donor and infused into patients must find their way through a long and winding network of veins, arteries and capillaries in order to home to the BM.
The purpose of this review is to present a description of the molecular and cellular cues that account for the exquisite specificity and efficiency of HSPC homing to their niches in the BM. Two questions that underlie both features of the homing process will be addressed: What molecular components and receptors are required for the different stretches of this voyage? What cues provide specificity for the BM? In the last part of this review some of the strategies that have been devised to improve HSPC homing and enhance the recovery of patients undergoing BMT will be discussed.
TOOLS FOR THE ROADLeukocyte trafficking has been extensively investigated and the major molecular components mediating their recruitment to inflammatory sites identified. In a seminal review from 1994, Timothy Springer proposed a "code" for leukocyte trafficking that was specified by the different components of a multistep process in which the combination of molecules involved in each step dictated the route followed by each leukocyte subset during physiology and disease(16).
Upon inflammation, signals emanating from the injured tissue induce the expression of receptors in the endothelium that allow leukocytes to be recruited to that specific area. Molecules that are induced include endothelial selectins, receptors of the immunoglobulin superfamily and chemokines. Recruitment is initiated by high-affinity but short-lived interactions between leukocytes and the endothelial cells lining the inflamed vessels(17). These interactions, mediated by P-and E-selectins, result in a rolling motion that allow leukocytes to become further activated. Selectins recognize highly glycosylated ligands on the surface of leukocytes(18). P-selectin glycoprotein-1 (PSGL-1) is the main, if not only, physiological ligand for P-selectin(19,20). PSGL-1 is also a physiological ligand for E-selectin(20,21), but in this context it acts in coordination with other glycoproteins, such as CD43(22,23), CD44(24) and E-selectin-ligand 1 (ESL-1)(25,26).
Although ligation of selectin ligands can result in signaling and leukocyte activation, it is the binding of small and basic cytokines present on the endothelial luminal surface, termed chemokines, that triggers potent signals which result in integrin activation(27). Chemokine signaling is mediated by G-protein coupled receptors (GPCR) that span the plasma membrane seven times. Integrins thus activated promote firm adhesion to their endothelial ligands, particularly vascular cell adhesion molecule (VCAM)-1 and intercellular cell adhesion molecule (ICAM)-1(27,28). Integrins are heterodimeric proteins comprised of α and β subunits of which two subfamilies, β1 and β2, are predominantly expressed on leukocytes and can be activated upon chemokine induced signaling(28,29). Other molecules that do not belong to either of these receptor families, such as CD44, also play a role in leukocyte and HSPC trafficking, and will be discussed below.
INITIAL INTERACTIONS WITH THE BONE MARROW MICROVASCULATUREHSPC trafficking is thought to essentially be similar to the process by which leukocytes migrate to inflamed tissues (Fig. 1). Like leukocytes, HSPC initiate their migration to the BM through transient and weak interactions with endothelial selectins(30-34). A feature of the BM microvasculature is that it constitutively expresses endothelial selectins, the integrin counter-receptor VCAM-1 and the chemokine CXCL12 which is displayed on its luminal surface(29,35,36). Discrete areas of HSPC or leukemic cell entry in the BM have indeed been shown to display strikingly restricted expression of CXCL12 and E-selectin in vivo(37). This remarkable characteristic is critical in allowing the trafficking of HSPC as well as other leukocyte subsets to the BM.
, where they establish contacts (niche contacts) that allow their long-term engraftment in the vascular, endosteal and CAR niches. Egress of HSPC from the BM occurs physiologically or after injection of certain drugs (such as G CSF) by disruption of their interactions with the niche (small arrows). Controversies and alternate uses of molecules at different steps of this scheme are described in the text. Circled numbers identify specific processes that have been targeted in order to manipulate HSPC trafficking (both homing and mobilization). These are listed in Table I and discussed in the text. END, endothelial cells; OB, osteoblasts.")
Steps of stem cell homing to the bone marrow niches. The BM microvasculature constitutively expresses endothelial selectins, integrin counter receptors and the chemokine CXCL12. This allows HSPC circulating in the blood to initiate labile interactions and a rolling motion that are mediated by endothelial selectins and their ligands. As they roll, binding of CXCL12 present on the luminal surface of endothelial cells activates adhesive receptors on HSPC that result in their firm arrest and locomotion in search of areas suitable for transendothelial migration. Once in the marrow cavity, HSPC navigate in search of niches guided by CXCL12-dependent and independent signals (big arrows), where they establish contacts (niche contacts) that allow their long-term engraftment in the vascular, endosteal and CAR niches. Egress of HSPC from the BM occurs physiologically or after injection of certain drugs (such as G CSF) by disruption of their interactions with the niche (small arrows). Controversies and alternate uses of molecules at different steps of this scheme are described in the text. Circled numbers identify specific processes that have been targeted in order to manipulate HSPC trafficking (both homing and mobilization). These are listed in Table I and discussed in the text. END, endothelial cells; OB, osteoblasts.
HSPC express high levels of ligands for both P-and E-selectins(30-32), which initiate rolling through interactions with these molecules(32,34). Inhibition or absence of both selectins results in a strong reduction in both HSPC rolling on the BM microvasculature and homing to the BM(32-34,38). It should be noted, however, that human CD34+ cells (the subset that contains most HSPC) appear to be more dependent than murine HSPC on selectin-mediated interactions to enter the BM(34). While P-selectin is the major player in initiating these interactions, E-selectin is required to allow slow rolling and activation of the adhesive machinery(39). In vivo studies showed that PSGL-1 expressed on HSPC functions as the main ligand for both selectins(38); the same study also demonstrated that additional ligands were required for the interactions with E-selectin. These additional ligands have not been characterized, but studies with human CD34+ cells and murine neutrophils suggest that CD44 may be one such ligand(24,40). Because CD44 is also a receptor for hyaluronan (HA) on activated T lymphocytes and HSPC(41,42), and HA is expressed on the surface of the BM microvasculature(43), it is conceivable that it functions as a dual receptor for both E-selectin and HA during HSPC homing.
While selectins are clearly important in mediating these initial rolling interactions, activated integrins can also mediate leukocyte rolling(44,45). In studies aimed at assessing the contribution of α4 integrins in HSPC homing, we found that simultaneous blockage or absence of these integrins and E-selectin almost completely prevented entry of HSPC into the BM(38). Expression and function of the α4β1 (or VLA-4) integrin has been extensively reported in both human and murine HSPC(46,47). We were surprised, however, to find that a second α4 integrin, α4β7, was also expressed by murine HSPC(48). The integrin α4β7 and its primary counter receptor, mucosal addressin cell adhesion molecule-1 (MadCAM-1), are involved in the entry of T lymphocytes to intestinal lymphoid organs(49). Our studies demonstrated that both α4β7 and MadCAM-1, but not α4β1, were involved in mediating rolling interactions with the BM microvasculature(48). Whether this pathway is also used by human HSPC remains undetermined. The redundancies revealed by these studies, where multiple adhesive ligandreceptor pairs appear to mediate the initial interactions of HSPC, support the notion that this step is critical for the specificity and efficiency of the homing process.
TURN SIGNALS: CXCL12The chemokine CXCL12 is an essential component of the hematopoietic niche(50). During homing, it is thought to provide the signals that indicate to an HSPC that it has arrived at the "target" organ and that it must take a turn at that specific location. Contrary to most chemokines, CXCL12 signals exclusively through one receptor, CXCR4. Although HSPC express other chemokine receptors, CXCR4-mediated signaling triggers the most potent physiological responses in these cells(51-53). CXCL12 is one of the most extensively studied chemokines, due in part to the dramatic phenotype of both CXCL12-and CXCR4-deficient mice and the involvement of CXCR4 in HIV infection(54-56). Studies in these mutant mice demonstrated an essential role for this signaling axis during hematopoiesis, including B lymphocyte development and colonization of the BM by HSPC and myeloid progenitors during ontogeny(57). A number of experimental observations further support the critical function of CXCL12 during hematopoiesis. First, it is expressed in the very cells that form the niche in which HSPC reside in the BM and contributes to their retention in those niches(6,29,58). This contribution was further evidenced in experiments where reduction of CXCL12 levels triggered by the cytokine G-CSF resulted in egress of HSPC into the peripheral circulation(59-61). Secondly, inhibition of the CXCL12/CXCR4 axis using antibodies(52) or pertussis toxin(62), or by genetic deletion(63), prevented HSPC homing to and engraftment in the BM.
At the mechanistic level, CXCL12 is thought to control homing by enhancing the adhesiveness of HSPC. At least in T lymphocytes, this is first achieved by the induction of microvilli collapse on the cell surface to allow for a larger area of cell contact with the endothelium(64). In human CD34+ cells, CXCL12 promotes a rapid and strong increase in the affinity and avidity of the β1 integrin α4β1 for its ligands, VCAM-1 and fibronectin(29,65). The functions of other adhesion receptors on HSPC, including α5β1(28), αLβ2(29), α4β7(48) and CD44(43) can also be modulated by CXCL12. These findings support a model in which CXCL12 exposed on the luminal side of the BM microvasculature(66) is detected by CXCR4 during HSPC rolling. Signals delivered by CXCR4 in turn activate integrin and non-integrin receptors on HSPC and promote their firm attachment to the BM vasculature(28,29).
Of the adhesive receptors modulated by CXCL12, β1 integrins in particular appear to play an essential role in HSPC homing to adult hematopoietic organs. Although β1-deficient HSPC have a normal capacity to differentiate into mature leukocytes in vitro, they are unable to colonize either the fetal liver during embryonic life or the BM of adult mice(67,68). A similar phenotype was observed in chimeric mice and in mice conditionally deficient in the α4 subunit of β1 and β7 integrins(69,70). Together, these experimental data suggest that CXCL12 and the α4β1 integrin act in coordination and are critical to mediate the arrest of HSPC on the BM microvasculature(29,48,71).
CROSSING THE FENCEWhile the adhesive pathways promoting the initial interactions of HSPC with the BM microvasculature have been well studied, those mediating the movement on and through the endothelium remain less well characterized. Insights into these processes have come from in vitro studies that analyzed leukocytes moving on and transmigrating through cultured endothelial cells. Recent studies have shown that leukocytes "palpate" the endothelium while they move in search of areas of transmigration(72). This movement, termed locomotion, is mediated by β2 integrins on monocytes(73) and requires the active formation of sensing protrusions on the leukocyte(72). On the endothelial side, this process is paralleled by the formation of "docking" structures that are enriched in both integrin counter-receptors (VCAM-1 and ICAM-1) and in the tetraspanins CD9 and CD151(74,75). Transendothelial migration of mature leukocytes can then occur between or through endothelial cells (referred to as the paracellular or transcellular routes, respectively), and it is mediated by homophilic as well as heterophilic interactions via receptors such as ICAM-1, platelet endothelial cell adhesion molecule (PECAM)-1, CD99, VE-cadherin, endothelial selective adhesion molecule (ESAM), and the junctional adhesion molecules (JAM)-A, B and C(76,77). Partial contributions of PECAM-1, CD99 and VE-cadherin have been described during the transmigration of human CD34+ cells through BM endothelial cells using in vitro systems(78-80). There is still a considerable gap in our understanding of the mechanisms of HSPC transmigration through the microvasculature of the BM. It is believed, however, that signals triggered by the chemokine CXCL12 are required for the diapedesis of HSPC across the BM vasculature.
What are the signals triggered by CXCL12 that are required for HSPC homing? One family of molecules activated by CXCR4 signaling has been thoroughly studied in the context of HSPC homing and engraftment. Rac1 and Rac2 belong to the Rac subfamily of Rho guanosine triphosphatases, and display a wide and hematopoietic-restricted pattern of expression, respectively(81). They play important roles in cytoskeletal reorganization, proliferation and survival of HSPC(82). In vitro migration of HSPC in response to CXCL12 was abolished in the absence of both Rac proteins(83). Although mice deficient in either protein display distinct hematopoietic defects, only the absence of Rac1 resulted in a partial deficiency in HSPC homing to the BM and an abnormal HSPC localization within the medullary space, and these deficiencies were further accentuated when both Rac proteins were absent(84). Since Rac proteins are important in the formation of lamellipodia and membrane ruffling in response to exogenous factors, these studies demonstrated redundant and important roles for Rac1 and Rac2 in integrating signals that induce HSPC motility in response to CXCL12(83,84). Associated with the function of Rac proteins, the guanine exchange factor Vav1 controls adhesive (e.g., α4 integrin activation) and chemotactic responses to CXCL12(85,86). Upstream of Rac1, cyclic AMP levels can control transendothelial migration and motility through modulation of CXCR4 levels on the cell surface. This mechanism, which also requires the (PKCζ)(87,88), may be important in all other steps of the homing process that involve CXCL12. In addition, HSPC express a number of receptors for other biological mediators such as lipids (sphingosine-1-phosphate)(89), chemoattractants (C3a)(90), nucleotides (UTP)(91) and neurotransmitters (dopamine, epinephrine or norephinephrine)(92) that can potentiate their transendothelial migration and adhesion through modulation of CXCR4-mediated signaling. Downstream of CXCR4 signaling, in vitro studies have shown that HSPC stimulated with CXCL12 or with cytokines produce matrix metalloproteases (MMP2 and MMP9) that facilitate their migration across basement membranes(93,94) and may also aid in the navigation through the marrow parenchyma(95). These observations suggest that transendothelial migration of HSPC is a tightly regulated process that results from the confluence of multiple signaling and adhesive pathways(77). Whether this process is similar to that described for mature leukocytes, and what is the contribution of the specialized BM endothelium, remain unanswered questions(96).
REACHING THE MARROWAre HSPC that cross the BM vasculature at the end of the road? The marrow space is not a homogenous tissue. As indicated at the beginning of this review, HSPC reside in very specific niches in close contact with specific cells (Fig. 1). It has been known for more than 30 years that pluripotent HSPC reside in or near the endosteal surface(97,98), but only recently have we learned of the remarkable capacity of HSPC to migrate to and engraft in these areas of the marrow [now defined as a distance of 12 or fewer cell diameters from the bone surface(99)] within just few hours of intravenous injection(99-101). These important observations indicated not only that HSPC can specifically home to the BM from the circulation, but that they can also search for a specific and appropriate location within a solid organ. Although the "rules of the road" must change when moving from a fluid to a solid milieu, it is likely that similar cues govern these final stages of homing.
A major problem in dissecting this process has been that the BM is a difficult organ to visualize. Pioneering studies by the group of Ulrich von Andrian, however, have benefited from multiphoton microscopy technology to image the dynamic movement of T lymphocytes within the BM parenchyma(102). These innovations have not yet been applied to the study of HSPC migration inside the BM, but prove that with the help of current technologies we may be able to understand this process with greater detail in the near future.
Receptors involved in intramedullary navigation include some that are also required during previous stages of homing (CXCR4 and β1 integrins), together with receptors highly specialized in guiding HSPC within the BM. A central role for the CXCL12/CXCR4 pathway in this process was suggested from studies showing that the hypoxic conditions of the endosteal region lead to CXCL12 expression, and favor the preferential tropism of HSPC to these regions of the marrow(103). Furthermore, inhibition or absence of Rac2 results in the inability of HSPC to engraft transplanted mice without affecting their capacity to enter marrow(84), suggesting that CXCR4 modulation of Rac2 activity is specifically required for intramedullary navigation. Modulation of CXCR4 signaling through other receptors (e.g., lipids, chemoattractants, nucleotides or neurotransmitters)(89-92) likely have an impact on HSPC movement inside the marrow as well, although this has not been formally demonstrated.
Evidence that β1 integrins participate in the navigation of HSPC within the BM parenchyma has been gained by the observation that specific inhibition of the α5 and α6 subunits (VLA-5 and VLA-6, respectively) results in reduced homing(104-106). While the ligand for VLA-5, fibronectin, is widely expressed, that of VLA-6, laminin, is restricted to the subendothelial basement membrane and BM parenchyma and absent from the endothelial surface(107). This suggests that VLA-6 function is restricted to intramedullary migration. In addition, blockade of α6 integrins in mice does not result in HSPC dislodgment from the BM(105). This contrasts with the effect of α4-and α5-integrin blocking reagents(106,108,109), and indicates that VLA-6 does not mediate interactions of HSPC with their niche.
Several receptors and matrix elements not involved in other stages of homing have recently been identified as critical in guiding the movement of HSPC within the marrow space. One of these is a calcium-sensing receptor (CaR) that can detect gradients of this cation. CaR is expressed on HSPC and guides its migration to the endosteal region, where Ca2+ levels are approximately 20 times higher than those found in the blood(110). Studies in CaR-deficient mice showed that this receptor was not required for homing into the marrow space, but rather to properly position HSPC near the bone surface, in the vicinity of osteoblasts(111). A similar role has been proposed for osteopontin (OPN), a matrix component produced by osteoblasts and deposited on the endosteal surface(112,113). Studies in OPN-deficient mice suggested that engagement of OPN by VLA-4 or CD44, two of its receptors on HSPC, contributes to the intramedullary navigation of HSPC towards the niche areas(112,113). Interestingly, another ligand for CD44, hyaluronan, was found to be expressed on the surface of HSPC and also affected the distribution of homed cells(114). Altogether, these observations suggest that the initial events of homing, i.e. interactions with the BM microvasculature and transendothelial migration, and the later events within the marrow are mediated, at least in part, by a different repertoire of receptors.
CUDDLING IN THE NICHEAs mentioned earlier, several niches for HSPC exist in the BM. HSPC can associate with osteoblasts (the endosteal niche)(3,4), endothelial cells (the vascular niche)(5) or reticular cells (CAR cells)(6). Which of these niches do HSPC chose? Although it is clear that they preferentially migrate near the endosteal surface, only a small fraction of them actually contact the bone interface(5,100). This would suggest that more than one type of niche can attract HSPC. If this is true, it remains to be determined whether this positioning is a stochastic process (i.e., HSPC stay in the first niche that they find) or rather that it depends on the status of each migrating HSPC (e.g., cell cycle or differentiation profile).
HSPC that successfully arrive at their niche can then establish interactions with cellular and extracellular elements that control their survival (SCF, CXCL12, osteopontin)(112,113,115,116), proliferation/quiescence (SCF, Angiopoietin-1, Ncadherin)(3,95,117), differentiation (Notch1)(118) or retention (VCAM-1, CXCL12)(6,60,109,119). The efficiency of HSPC engraftment in this niche is illustrated by the productive establishment of long-term hematopoiesis with daily release of billions of blood cells in virtually every transplanted recipient. Interestingly, the interactions of HSPC with their niche appears to be reversible since the disruption of either the CXCL12/CXCR4 or α4β1/VCAM-1 pathways in adult mice results in a dramatic release of HSPC from the BM into the circulation(6,70,71,120).
As HSPC differentiate into different lineages, it is believed that increasingly mature cells relocate closer to the vascular areas, ready to enter the circulation(98). How this movement occurs is largely unknown, with few exceptions: Megakaryocytic progenitors are often found in contact with BM vascular sinusoids where they continuously release platelets into circulation. Two chemokines, CXCL12 and fibroblast-growth-factor 4, were shown to mediate megakaryocyte recruitment to a vascular niche thus promoting platelet production(121). This observation exemplifies that different cues can guide the migration of distinct stem and progenitor cells to their respective niches within the BM.
REPAVING THE ROAD: STRATEGIES TO IMPROVE HSPC TRAFFICKINGDetailed characterization of the "rules of traffic" followed by HSPC as they migrate to their niches will be tremendously beneficial. One very direct application for hematologists is the possibility to manipulate HSPC homing to their advantage. In the context of BMT, a major goal is to improve the process of homing to allow for faster recovery of hematological parameters in transplanted patients. It might also allow to enhance the engraftment of genetically manipulated HSPC(122). One particularly promising application is to improve the homing of cord blood (CB)-derived HSPC. CB represents an unlimited source of HSPC for BMT(123), but because the amount of blood obtained from one placenta is small (~100-200 milliliters) and the number of HSPC collected low, its use has been limited to patients of low weight and to children(124,125).
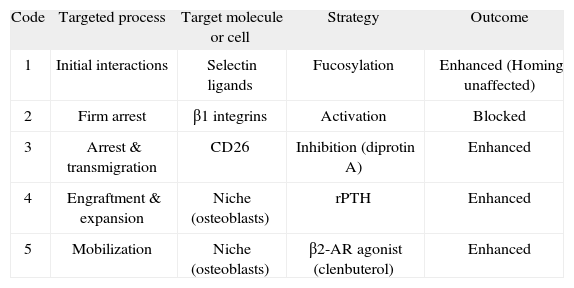
All the steps identified during the homing process can potentially be targeted in order to enhance HSPC engraftment. I will briefly review now strategies developed over the last few years that attempted to manipulate molecules at various steps of the homing process, and that have attained varying degrees of success (Figure 1 and Table I).
Manipulation of HSPC homing to the bone marrow
| Code | Targeted process | Target molecule or cell | Strategy | Outcome |
| 1 | Initial interactions | Selectin ligands | Fucosylation | Enhanced (Homing unaffected) |
| 2 | Firm arrest | β1 integrins | Activation | Blocked |
| 3 | Arrest & transmigration | CD26 | Inhibition (diprotin A) | Enhanced |
| 4 | Engraftment & expansion | Niche (osteoblasts) | rPTH | Enhanced |
| 5 | Mobilization | Niche (osteoblasts) | β2-AR agonist (clenbuterol) | Enhanced |
Description and outcome of the different strategies devised to enhance HSPC homing. Code numbers correspond to the targeted steps identified in Figure 1. Details of these strategies are provided in the text. rPTH, recombinant rat parathyroid hormone; β2-AR, beta 2 adrenergic receptor.
Because this process is mainly mediated by endothelial selectins, we and others attempted to enhance HSPC interactions with the vascular bed of the BM by enhancing the expression of selectin ligands on HSPC(126,127). Initial experiments using CD34+ cells derived from CB samples demonstrated that, compared to those derived from mobilized peripheral blood or BM, these cells initiate rolling on the BM microvasculature of NOD/SCID mice (a common murine model to study human hematopoiesis) with low efficiency(34). This reduction was due to the lack of expression of ligands for both P-and E-selectins on about one third of CB-derived CD34+ cells. Biochemical and gene expression analyses demonstrated that one type of enzyme involved in the synthesis of selectin ligands was markedly reduced in these cells. This enzyme, a fucosyltransferase (FuT), acts in concert with other glycosyltransferases to properly modify scaffold proteins (PSGL-1 and CD44) and to generate functional selectin ligands(127). To artificially induce selectin ligands on CB-derived CD34+ cells, a recombinant soluble form of FuT6 was added to the cell media, together with the appropriate co-factors and a fucose donor (GDP-fucose). After a brief incubation at 37°C, this treatment induced a robust expression of ligands for both P-and E-selectin in all CD34+ cells. In addition, treated cells could more efficiently roll on and adhere to the BM microvasculature of NOD/SCID mice in vivo(127). Thus it was surprising and disappointing that this did not result in an increased number of cells that homed inside the BM. Conflicting results from one group suggested, however, that long term engraftment could be enhanced by fucosylated CB-CD34+ cells(126). It is currently difficult to reconciliate both observations, which might be due to minor technical variations. One potential interpretation of why increased interactions with the vasculature did not translate in enhanced homing is that a different set of molecules required for homing (e.g., chemokine receptors or signaling proteins) act as a bottleneck for the entry of CB-HSPC inside the BM. Further research to unveil these limiting molecules will be important to get a full benefit from this strategy.
Enhancing the firm arrest of HSPC on the BM vasculatureβ1 integrins are essential for the firm arrest of HSPC on the BM microvasculature(29). Because cultured HSPC displayed a reduction in β1 integrin-mediated binding, a strategy was devised to increase these interactions using a monoclonal antibody that locks β1 integrins in a high affinity state. Interestingly, this treatment resulted instead in the complete inhibition of HSPC homing and engraftment(128). In retrospect, this was perhaps an expected outcome. Integrin function in HSPC interacting with the BM microvasculature is controlled by CXCL12, and is characterized by a rapid and transient increase(28,29,65). This allows for strong binding to the vasculature after rolling, followed by release of these bonds to allow further movement on and across the endothelium. Locking β1 integrins in a high-affinity state, it seems, favored the initial firm interactions but prevented the subsequent migration of HSPC. This strategy provides an example of how a biological phenomenon must be carefully characterized before it can be successfully manipulated.
Modulating the CXCL12/CXCR4 axisAs evidenced by the studies described above, CXCL12-triggered signaling is essential for HSPC homing. It controls multiple steps of this process, including firm arrest, transendothelial migration and intramedullary navigation in search of niches in the BM. Thus, a great deal of effort has been put into finding ways to enhance CXCR4-mediated signaling in HSPC. For example, upregulation of CXCR4 expression on human CD34+ cells by cytokines (SCF and interleukin-6) resulted in enhanced migration towards CXCL12 in vitro and engraftment of NOD/SCID mice(52). Alternatively, treatment of human or murine HSPC with lipids or chemoattractants (S1P and C3a, respectively) potentiated the responses mediated by CXCR4 in vitro and enhanced engraftment in vivo(89,90). Likewise, treatment of murine or human HSPC with factors present in the serum of G-CSF-mobilized blood enhanced their responses to CXCL12 and homing by promoting CXCR4 incorporation into physiological signaling platforms (lipid rafts)(129).
A markedly different and elegant strategy was described by the group of Hal Broxmeyer. Their approach was based on the specific inhibition of CD26, a serine dipeptidylpeptidase (DPP-IV) expressed on the surface of hematopoietic cells. CD26 cleaves the N-terminal dipeptide from CXCL12, thus rendering it inactive(130). In the context of homing, CD26 present on the surface of rolling HSPC is thought to inactivate CXCL12 presented by the BM endothelium, thus reducing the levels of functional chemokine over time(130,131). In vitro studies demonstrated that absence or blockage of CD26 prevented CXCL12 inactivation; in vivo this translated into a remarkable enhancement of homing and engraftment of both murine and human HSPC(131,132). This approach is probably the most promising attempt to manipulate HSPC homing to date, and serves to illustrate that this is an attainable goal that may soon benefit the clinic.
Targeting the stem cell nicheAt the end of the homing process, the interactions established between HSPC and their niche favors their longterm preservation and function. The group of David Scadden hypothesized that targeting the osteoblastic cells that constitute the endosteal niche might enhance the engraftment and survival of transplanted HSPC. They took advantage of their observation that expression of a constitutive active form of the parathyroid hormone (PTH)/PTH-related peptide receptor in transgenic mice, or treatment with exogenous PTH led to an expansion of the HSPC pool(4). They thus treated mice repeatedly exposed to chemotoxic agents (cyclophosphamide) or lethally irradiated mice after BM transplantation with exogenous PTH. This treatment resulted in protection of HSPC from cytotoxic damage as well as in an expansion of the transplanted HSPC(133). The potential benefits of this strategy in the clinic are enormous, not only in the context of BMT, but also in the outcome of leukemic patients undergoing chemotherapeutic regimes.
Enhancing the mobilization of HSPCHSPC egress from the BM can be enhanced by treatment with a number of agents, a process termed mobilization, and is generally considered to be caused by disruption of retention signals in the BM niches(134). For this reason this process is envisioned as a reversal of the last steps of homing. Mobilization is a process of considerable clinical interest since the vast majority of autologous or allogeneic BMT procedures today use HSPC obtained from mobilized donors due to the increased number and superior engrafting capabilities of these cells compared to those from BM or CB units(135).
Although many of the cellular and molecular participants of this process are still unknown, most of the signaling and adhesive molecules so far involved in HSPC mobilization are also required for their homing(134). In particular, blockage of both the VLA-4/VCAM-1 and CXCL12/CXCR4 pathways either directly [with antibodies or antagonists(60,109,136,137)] or indirectly [by cytokine-induced downregulation of gene expression or protease-mediated degradation(59,61,119)] have been extensively studied as strategies to enhance HSPC mobilization. One example of the potential utility of such strategies is the case of AMD3100. This bicyclam is a designed CXCR4 antagonist(138) that induces HSPC mobilization by itself or by synergizing with G-CSF(137), and is currently being tested in several clinical trials.
A completely different approach to enhancing HSPC mobilization was provided recently by our group. We found evidence that signaling through the sympathetic nervous system (SNS) was required for G-CSF to downregulate niche activity and to trigger mobilization. Correspondingly, simple co-injection of a β2-adrenergic receptor agonist (clenbuterol) enhanced cytokine-induced mobilization(61). These findings suggest that pharmacological manipulation of the SNS can be clinically useful to increase HSPC mobilization (using adrenergic agonists) or, conversely, to enhance their engraftment in the BM niches (using adrenergic antagonists)(61,139).
FUTURE AVENUESThe observations accumulated during the study of the mechanisms of HSPC trafficking will have an impact in several areas of stem cell biology, most importantly perhaps in tissue regeneration and cancer: HSPC accumulate in areas of damage and appear to contribute to tissue repair(140). Likewise, BM-derived HSPC migrate to pre-metastatic lesions and may contribute to tumor spread(141). As in the case of homing to the BM, both of these processes seem to require the CXCR4/CXCL12 and VLA-4/VCAM-1/fibronectin
pathways(71,103,141). Given the striking similarity between all these processes, it is tempting to speculate that the trafficking of stem and progenitor cells in search of certain niches follow the same basic rules described here. Finding strategies to potentiate HSPC recruitment to ischemic or damaged tissues, or to block it when primary tumors are detected will be major challenges for the future.
Another area of great interest will be to understand whether the homing of pathogenic stem cells to "their niches" differs from that of normal stem cells, and if so how can we take advantage of those differences. This last point is prominently illustrated by the recent finding that chronic myelogenous leukemia-initiating cells (a kind of leukemic stem cell) rely on CD44 as a BM homing receptor more heavily than normal HSPC do, and that blockage of CD44 partially protects animals from this type of leukemia(142). As more leukemia and solid tumor types that rely on founding cancer stem cells are being described(143,144), it will become a priority to characterize their migratory patterns.
Since the pioneering description in 1956 of cells from a donor that could "in special circumstances, not merely survive and multiply in another, but even replace the corresponding cells of the host and take over their functions"(145), a long and exciting road of discoveries in the biology of hematopoietic stem cells was initiated that has led to enormous medical benefits and will continue to guide us to future challenges in the biomedical research of the twenty-first century.
CONFLICT OF INTERESTThe author declares no financial conflict of interest.
I want to express my gratitude to Linnea Weiss, Daniel Lucas-Alcaraz and Weiming Kao for helpful comments and corrections during the writing of this review. The author is supported in part by a Scientist Development Grant from the American Heart Association.