


La fosforilación es una modificación post-traduccional (PTM) reversible, la cual juega un papel crucial en cascadas de señalización, controlando muy diversas respuestas biológicas, tales como el crecimiento y la diferenciación celular, y los procesos de invasión, metástasis y apoptosis(1-3). Más aún, la identificación de residuos fosforilados implica poder profundizar y entender diferentes mecanismos de transducción de señales. Además, la activación y desactivación mediante fosforilación de las cascadas de señalización, es relevante en el desarrollo de patologías como el cáncer(4,5).
En esta revisión se exponen, discuten e ilustran algunas de las innovaciones proteómicas recientes y útiles para abordar estudios de investigación clínica, especialmente aquellos cuyo objetivo sea la identificación de proteínas fosforiladas a nivel funcional.
Se detallará el fundamento y utilidad de diferentes cromatografías basadas en el uso de metales para enriquecer en péptidos fosforilados, junto con los distintos tipos de cromatografías de fase reversa para la limpieza de sales y concentración de los mismos. Más aún, se detallan posibles estrategias basadas en Espectrometría de Masas (MS) para la identificación de los residuos fosforilados.
Phosphorylation is a reversible post-translational modification which plays a crucial role in the regulation of signalling pathways; therefore it controls many biological responses including cell growth and differentiation, as well as invasion, metastasis and apoptosis.
Current phosphoproteomic approaches are a powerful tool for monitoring global molecular responses to the activation or deactivation of signal transduction pathways. Moreover, relevant advances in Proteomics and Mass Spectrometry (MS) technologies have enabled the identification of thousands of known and novel phosphorylation targets. Indeed, today's scientists take heed of those strategies for functional phosphoprofiling studies of signalling pathways, for drug discovery and for the understanding of the etiology of different diseases.
La fosforilación es una modificación post-traduccional ("posttransductional modification", PTM) reversible, que es clave en los procesos biológicos de organismos procariotas y eucariotas. Los procesos celulares, como la adaptación a los cambios del entorno celular, están regulados por la fosforilación y defosforilación de proteínas (por ejemplo, quinasas). En organismos procariotas esta PTM ocurre principalmente en residuos de histidina, ácido glutámico y ácido aspártico; sin embargo en eucariotas ocurre en serinas, treoninas y tirosinas.
La fosfoproteómica consiste en el estudio de la fosforilación de las proteínas que forman parte de la célula o tejido en un estado determinado. Los abordajes fosfoproteómicos actuales permiten realizar estudios de fosforilación tanto de una como de muchas proteínas(6).
Debido a que los proteomas celulares consisten en muchas y diferentes proteínas, de las cuales, las fosforiladas representan un tercio, es realmente difícil identificar todos los residuos fosforilados de una muestra compleja(7-10). Por tanto, uno de los objetivos más laboriosos en las aplicaciones de las técnicas fosfoproteómicas, consiste en la extracción de todas las proteínas fosforiladas de los lisados celulares y/o tejidos con el fin de identificar fosfoproteínas cuyo nivel de expresión sea bajo. Por ejemplo, se estima que los receptores Arsandrógenoson fosforilados en diferentes residuos, y tan sólo algunos de ellos son conocidos. Además, las quinasas que median dichas fosforilaciones tampoco han sido aún establecidas en su totalidad. Chen y cols. realizaron un estudio con el fin de elucidar el papel de la fosforilación en relación al cáncer de próstata(11). Esto implica gran dificultad y procesos tediosos, ya que dicha modificación posttraduccional es una reacción reversible y altamente dinámica, la cual además, puede ocurrir en varios residuos de una misma proteína(12).
Los abordajes fosfoproteómicos actuales constituyen una herramienta muy poderosa para estudiar respuestas celulares tanto a nivel global (fosfoproteomas) como a nivel individual de una determinada proteína. Además, el gran avance de la espectrometría de masas (MS) ha permitido identificar y cuantificar miles de residuos fosforilados(13). Por estos motivos, hoy en día muchos científicos recurren a estrategias proteómicas y de espectrometría de masas para la identificación de residuos fosforilados correspondientes a proteínas que forman parte de cascadas de señalización. La razón última de esto, es que el estudio de la activación y desactivación de dichas cascadas mediante fosforilación, puede ayudar a desarrollar nuevos fármacos y permite profundizar en los mecanismos de las diferentes patologías. Recientemente, estudios relacionados con diferentes tipos de cáncer (por ejemplo en leucemia mieloide aguda)(3,14) señalan como futuras perspectivas la importancia de la identificación de las proteínas fosforiladas y defosforiladas durante el comienzo y desarrollo de la enfermedad para un mejor entendimiento de la propia patología. Sin embargo, para entender los complejos eventos biológicos que ocurren en las células, es necesario identificar las quinasas que forman parte de las cascadas de señalización, y determinar si éstas están activadas o desactivadas mediante fosforilación. Estos estudios complejos requieren además de la identificación de las fosfatasas implicadas en dichos procesos biológicos(15).
MÉTODOS ÚTILES PARA EL ESTUDIO DE LA FOSFORILACIÓN EN PROTEÍNAS Y PÉPTIDOSMarcajes radioactivos (P32)El uso de la radioactividad supuso un importantísimo avance químico y biomédico ya que permitió marcar las proteínas mediante la adición de isótopos radioactivos. El marcaje con P32 suele utilizarse en combinación con los geles 2DE(16), y permite cuantificar el aumento o disminución de las proteínas marcadas bajo distintas condiciones (ejemplo, estrés y estímulo). En el marcaje radioactivo se marca una proporción baja de la proteína y no es una tecnología óptima para estudios a gran escala, ya que mide la incorporación del marcaje y no del nivel de la fosforilación. Por tanto, existe el riesgo de que proteínas con bajo nivel de expresión no sean marcadas radioactivamente, y que, por otro lado, no sean visualizadas en los geles 2DE. Por tanto esta metodología, aún siendo de gran utilidad, tiene el inconveniente de que es dependiente tanto de la tasa de recambio de las proteínas como también del nivel de expresión proteico.
Anticuerpos fosfoespecíficosExisten anticuerpos de unión a tirosinas, serinas y treoninas fosforiladas, los cuales, en combinación con ensayos de inmunoprecipitación, permiten enriquecer la muestra en fosfoproteínas a partir de la mezcla compleja de proteínas de un lisado celular. Se ha comprobado que el uso de algunos fosfoanticuerpos para inmunoprecipitar proteínas cuyo nivel de expresión es bajo, poseen un rendimiento no muy elevado y cierta inespecificidad. También, su uso en combinación con técnicas de inmunoblot permiten detectar si las proteínas a estudiar están o no fosforiladas(17).
Los anticuerpos que se unen a determinadas secuencias peptídicas fosforiladas (por ejemplo SerXThr, donde X es cualquier amino ácido, Ser y Thr corresponden a serina y treonina), pueden ser utilizados para inmunoprecipitar determinadas proteínas fosforiladas a partir de lisados celulares y/o tejidos. El material inmunoprecipitado puede ser visualizado mediante geles (SDS-PAGE, 2DE), y subsecuentemente las proteínas de interés serán digeridas mediante un enzima (generalmente tripsina) para su análisis posterior mediante MS(18). Esta vía permite por tanto enriquecer en sub-fosfoproteomas específicos.
Por otro lado, los anticuerpos que reconocen residuos fosforilados de manera independiente a la secuencia peptídica, son agentes de purificación mediante afinidad muy útiles. Este es el caso de los anticuerpos anti-tirosina, los cuales están siendo extensivamente usados(19). Esta estrategia es de gran relevancia, ya que la forma fosforilada en tirosinas ocurre en menor medida que la de serinas y treoninas. Además, se ha descrito que algunas tirosinquinasas juegan un papel importante en la patología del cáncer. Las proteínas fosforiladas en tirosina pueden ser inmunoprecipitadas a partir de mezclas complejas, las cuales pueden ser -tras la digestión tríptica-analizadas mediante MS utilizando modos de escaneo específicos(20). Del mismo modo, las fosfotirosinas inmunoprecipitadas, pueden ser sometidas a diferentes métodos de fosfo-enriquecimiento (se detallan a continuación en el siguiente apartado) y luego ser analizadas mediante MS(21).
Los anticuerpos anti-tirosina han sido recientemente y exitosamente utilizados por Zhang y cols.(19) para capturar péptidos fosforilados en dicho residuo. Este estudio permitió a este grupo identificar 180 sitios de fosforilación en tirosinas. Gygi y cols.(11) identificaron 385 residuos fosforilados partiendo de una concentración proteica mayor que la utilizada por Zhang y cols.(19).
Se han generado y utilizado exitosamente también anticuerpos anti-fosfoserina y anti-fosfotreonina. Sin embargo, actualmente estos anticuerpos no son extensivamente utilizados en estudios fosfoproteómicos. Esto es debido a que tienen una especificidad menor comparada con la de los anticuerpos anti-tirosina. Más aún, hay alternativas y métodos más eficientes para enriquecer en serinas y treoninas fosforiladas -por ejemplo IMAC y TiO2- que se detallan más adelante.
Técnicas de cromatografía de afinidad basadas en el uso de metales en micro-columna (IMAC y Dióxido de Titanio)IMAC ("Immobilized Metal ion Affinity Chromatography") y la cromatografía con dióxido de titanio (TiO2), emplean dos resinas útiles para el enriquecimiento en péptidos fosforilados previo al análisis por MS. En ambas cromatografías, el material o resina con la que se empaquetan las microcolumnas contiene metales y constituye la fase estacionaria. Esta fase estacionaria posee afinidad por grupos fosfato cargados negativamente.
a IMAC es una técnica de enriquecimiento de fosfopéptidos muy usada, basada en la unión de fosfopéptidos cargados negativamente a iones metálicos (Fe3+, Ga3+ o Co2+). La unión se establece por atracción electrostática entre cargas positivas y negativas.
Esta técnica ha sido usada extensivamente para el enriquecimiento de péptidos fosforilados y su detección por espectrometría de masas(13-19,23-30), ya que la incorporación de un fosfato a un péptido le hace ser más hidrofílico.
La muestra que contiene fosfopéptidos y péptidos no fosforilados, es disuelta en una solución ácida (fase móvil) antes de ser introducida en la fase estacionaria. El pH de elección suele ser de 2.7-2.9. Esta acidificación se desarrolla para promover interacciones entre los fosfopéptidos y la fase estacionaria, y para minimizar la unión de los péptidos no fosforilados cargados negativamente(26). En condiciones de pH ácido, el valor del pKa del grupo fosfato de los péptidos fosforilados es 1, y el correspondiente al de los grupo carboxilo es 4, de tal forma que los radicales fosfato estarán ionizados y con carga negativa, mientras que los grupos carboxilo estarán protonados. Así pues, a pH ácido, los fosfopéptidos cargados negativamente se unen a los iones metálicos cargados positivamente (Fe2+, Ga3+, etc) a través de interacciones electrostáticas y se minimizará la unión a péptidos ácidos. Sin embargo, los péptidos altamente ácidos competirán por la unión a los metales debido a su alto contenido en residuos de aminoácidos acídicos (ácido glutámico y ácido aspártico).
b La afinidad del dióxido de titanio (TiO2) por iones fosfato en solución acuosa fue comprobada por Connor en 1999(31). Pinkse y cols. en el año 2004 profundizaron en la capacidad del TiO2 para unirse a fosfopéptidos(21,32). Ellos vieron que, bajo condiciones ácidas (pH 2.9), al introducir el analito en una columna de TiO2 se promueve la unión de fosfopéptidos a dicha resina. Los fosfopéptidos eran subsecuentemente eluidos de la columna usando un tampón alcalino (pH 9.0). Posteriormente los fosfopéptidos eluidos eran concentrados en una columna de fase reversa (R3) y analizados por LC-MS/MS.
El dióxido de titanio empaquetado en micro-columnas en combinación con DHB (ácido 2,5 dihidroxibenzóico) en el proceso de elución aumenta la selectividad de retención de péptidos fosforilados, y la retención de péptidos ácidos no fosforilados disminuye, en comparación con el uso de ácidos como salicílico, benzóico, fosfórico, fórmico o acético. Parece ser que el mecanismo consistiría en que las moléculas de DHB compiten eficientemente con los péptidos no fosforilados para la retención a la superficie del TiO2, mientras que la unión de los fosforilados no se ve afectada. Por tanto hasta ahora el DHB es el ácido más eficiente para prevenir la retención de los péptidos no fosforilados mientras que retiene la habilidad del TiO2 para unir los fosforilados. El efecto del DHB en combinación con IMAC (Fe3+; otro sistema de enriquecimiento de fosfopéptidos) resulta en la unión de un número alto de péptidos no fosforilados(33), esto indica una mayor selectividad de unión de péptidos fosforilados en la micro-columna de TiO2 que con la resina de IMAC. El dióxido de titanio (TiO2) parece ser un sistema altamente prometedor, pero al igual que IMAC, aunque en menor medida, tiene el inconveniente aún por resolver, de la unión de péptidos ácidos no fosforilados. Este estudio comparativo se realizó con proteínas modelo como ovoalbúmina y caseína(33). Además, innovaciones recientes respecto al TiO2(34) demuestran que dicho material en presencia de ácido glicólico mejora aún más la retención de péptidos fosforilados. Más aún, Larsen(34) demuestra cómo el uso de diferentes detergentes que se utilizan habitualmente en la preparación de muestras, interfieren en mayor grado en el proceso de retención de fosfopéptidos al material IMAC que con respecto al material de empaquetamiento dióxido de titanio (TiO2).
Se ha demostrado que tanto IMAC como TiO2 poseen cierta inespecificidad por péptidos ácidos no fosforilados (por ejemplo por ácido aspártico y/o glutámico) y por péptidos donadores de electrones (por ejemplo, péptidos conteniendo histidina). Ficarro y cols.(21) solucionaron dicho problema esterificando los péptidos ácidos. De esta forma, los péptidos cargados negativamente solo serían los que tuvieran grupos fosfato. Sin embargo la esterificación resultó no ser eficiente al 100% en todos los péptidos ácidos y además complicó en gran medida la complejidad del espectro debido a las señales de esos péptidos con diferentes grados de esterificación. Por otra parte, en este caso la esterificación requería de liofilización de la muestra, la cual puede causar pérdidas de fosfopéptidos(35).
Técnicas de cromatografía de fase reversa en micro-columnaEstas técnicas simplifican el análisis de la muestra, y su uso de forma previa al análisis mediante MS es de gran utilidad para la limpieza de los péptidos de interés. Esto es debido a que enriquecen en función de la hidrofobicidad o hidrofilia del analito a estudiar. Por tanto la muestra se enriquecerá en analitos hidrofóbicos o hidrofílicos en función del material con que se empaqueten las microcolumnas.
En las micro-columnas de fase reversa, el analito es disuelto en una solución polar móvil antes de ser cargado en una fase estacionaria hidrofóbica. Por medio de interacciones hidrofóbicas, el analito se une a la fase estacionaria, que está formada por micro-esferas cubiertas de cadenas lineales de hidrocarbonos. La elución del analito de la fase estacionaria se consigue usando solventes hidrofóbicos, los cuales rompen las interacciones hidrofóbicas entre el analito y las cadenas de hidrocarbonos de la fase estacionaria. Normalmente el acetonitrilo es el solvente que se usa como eluyente.
El empaquetamiento de las micro-columnas con material R3 favorece el enriquecimiento en péptidos hidrofílicos(36,37) y simplifican la complejidad del analito. El grafito(38) también puede ser usado con el mismo fin en el caso de proteínas fosforiladas(39). Por otra parte, estudios recientes demuestran la utilidad del material C18 en abordajes para el estudio de la fosforilación(40). Más aún, Thingholm y cols.(41) demuestran que la combinación de R3 con C18 es altamente eficiente para la limpieza, eliminación de sales y concentración de fosfopéptidos que provienen de muestras complejas. Lo interesante de dicha combinación es que el material C18 empaquetado en el extremo de la micro-columna impide que el material R3 sea arrastrado en el proceso de elución de la muestra, lo cual deterioraría el análisis mediante LCESI. En dicho trabajo, se identifica un elevado número de fosfopéptidos demostrando la utilidad de dicha combinación para muestras biológicas.
ESPECTROMETRÍA DE MASAS (MS)La Espectrometría de Masas permite el análisis de la fosforilación, más aún, permite identificar los residuos fosforilados de las proteínas o péptidos a estudiar, aunque implica una gran complejidad.
La detección de residuos fosforilados -en serinas, treoninas y tirosinas en eucariotas- mediante MS no es igual en estos tres residuos. La fosforilación en serinas o treoninas suele ser lábil, mientras que en tirosinas, los fosfopéptidos son relativamente más estables. Esto implica estrategias diferentes en MS para su detección e identificación del sitio fosforilado(42).
Causas que dificultan la detección de la fosforilación mediante MSA pesar de ser una de las estrategias más relevantes hoy en día, la detección de la fosforilación mediante MS resulta complicada por 5 razones: (a) una estequiometría baja, es decir, la forma fosforilada de una proteína se encuentra en menor proporción que la no fosforilada; (b) los residuos de fosforilación de una proteína pueden variar, es decir, una proteína puede fosforilarse en diferentes residuos, lo cual puede implicar la activación o inhibición funcional de la propia proteína; (c) en la célula, el nivel de expresión de las proteínas que se fosforilan normalmente suele ser bajo, por eso el enriquecimiento es un prerrequisito para su estudio(33);
(d) un rango dinámico limitado, es decir, aunque se pueden detectar los residuos de fosforilación, los sitios minoritarios puede resultar de difícil detección; (e) las fosfatasas pueden desfosforilar los residuos, y por tanto el uso de inhibidores de fosfatasas es otro prerrequisito(43).
Por estos motivos, el estudio de fosfopéptidos por MS es un desafío por sí mismo, pero además los grupos fosfato de los péptidos están cargados negativamente y se cree que esto puede disminuir la eficiencia de la ionización en el análisis MS positivo. Esto implica una mayor dificultad en la detección de fosfopéptidos por espectrometría de masas comparado con péptidos carentes de dicha PTM(28). Otro inconveniente es la baja abundancia de éstos en comparación con los no fosforilados, por lo que los fosfopéptidos pueden quedar ocultos, haciendo muy difícil su detección en muestras altamente complejas. Por estas razones, el enriquecimiento de fosfopéptidos es prioritario y necesario para su análisis por MS.
Identificación de los residuos fosforilados mediante espectrometría de masasPara la identificación de los residuos fosforilados es necesario el uso de la espectrometría de masas en tandem (MSn) empleando ionización MALDI (Ionización, desorción mediante láser asistida con matriz, Matrix-Assisted Laser Desorption/Ionization)(44) y ESI (Ionización en electrospray, ElectroSpray Ionization)(45-47) que permiten realizar estudios de identificación de residuos fosforilados adecuadamente.
Cuando se buscan fosfopéptidos por espectrometría de masas, existen "huellas-fosfato" en el espectro de fragmentación (MSn) para la identificación del residuo fosforilado. A continuación se detallan dichas "huellas-fosfato"(28).
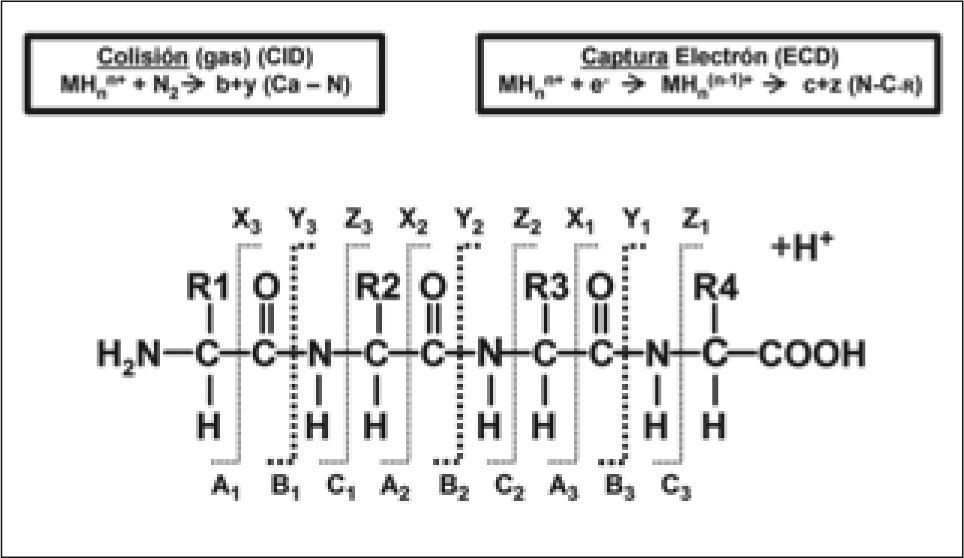
El grupo fosfato puede perderse de los residuos de serina y treonina por la reacción de β-eliminación formándose ácido fosfórico (H3PO4). Esto se debe a la fragmentación CID (Collision Induced Dissociation, colisión inducida por disociación)(36,48) y elución alcalina de los fosfopéptidos. En consecuencia, se produce la pérdida de 98Da (Da, Unidad de m/z) del residuo fosforilado (80Da del grupo fosfato y 18Da del agua), que es utilizado como una identificación positiva de fosfoserinas y fosfotreoninas.
Si el grupo fosfato se posiciona en un residuo de serina, aparecerá un pico 69Da inferior al fosfopéptido que sufrió la β-eliminación. Este pico aparece por la conversión de fosfoserina en dehidroalanina durante la β-eliminación. La presencia de 69Da y 167Da (98Da+69Da) son indicativos de una fosfoserina en la secuencia peptídica. La β-eliminación de fosfotreonina da lugar a la formación de ácido dehidro- 2-aminobutírico (83Da), el cual sirve de indicador de fosfotreonina.
La fosfotirosina, sin embargo, no sufre la β-eliminación debido a su cadena aromática. En su lugar, la fragmentación con CID causa que la fosfotirosina pierda H3PO (80Da). Además, a menudo se visualiza el ión imonio de la tirosina de 136Da en los espectros de péptidos que contienen tirosina. Un residuo fosforilado en tirosina resulta en un ión imonio de 216Da (136Da+80Da), el cual sirve de diagnóstico de fosfotirosina en la secuencia(49).
Estrategias para el análisis de los fosfopéptidos en espectrómetros de masas en tandem que incorporan ionización tipo ESI y un triple cuadrupoloExisten varias estrategias posibles para este análisis:
- a)
Mediante la selección en el modo MS de péptidos con pérdida neutra de 80–98Da (o 49Da si el péptido posee doble carga), para más tarde fragmentarlos y analizarlos en el modo MS/MS.
En un triple cuadrupolo (QQQ)(38,39,50,51), todos los iones entran en Q0 (transmisión), a continuación, se puede fijar Q1 para la detección de una masa de valor específica (80–98Da), tras lo que se produce su fragmentación (selección y fragmentación). Finalmente los iones "hijo" son analizados en Q2.
En un triple cuadrupolo acoplado a un analizador tipo "tiempo de vuelo" (QqTOF)(51), el proceso es similar. Todos los iones entran en Q0 (transmisión), a continuación, se puede fijar Q1 (selección) para la detección de una masa de valor específica (80–98Da). Esos iones seleccionados son enviados a Q2, que en este caso es la célula de colisión o fragmentación (CID). Finalmente los iones "hijo" son analizados en el TOF.
- b)
Mediante la selección de los 3 péptidos más intensos en el modo MS, para más tarde fragmentarlos y analizarlos en el modo MS/MS. El proceso es similar al descrito en el apartado anterior, salvo que en vez de seleccionar un valor de masa específico (80–98Da), se seleccionan en cada ciclo, los 3 péptidos de mayor intensidad en el modo MS, para posteriormente fragmentarlos y analizarlos en el modo MS/MS.
En ambos casos (a) y (b), la lista de masas obtenidas son introducidas en programas bioinformáticos (Mascot www.matrixscience.dk,Vemshttp://yass.sdu.dk/), donde se seleccionará como modificación variable la fosforilación en serinas, treoninas y tirosinas. En estos programas, los péptidos obtenidos como coincidentes, que incluyan una pérdida de 80–98Da serán susceptibles de estar fosforilados. La visualización de los espectros de fragmentación y la detección en ellos de patrones característicos de fosforilación, permitirán validar la existencia de dicha PTM en la muestra analizada.
La combinación de cromatografía líquida (LC) a este sistema, tiene un considerable valor para el análisis de fosfopéptidos, ya que la separación de los péptidos mediante LC disminuye la supresión tan comúnmente observada en el caso de fosfopéptidos(49). En este método, los péptidos son introducidos en columnas de diámetro pequeño (nanocolumnas de 75μm diámetro interno) que contienen material C18, para eliminar impurezas. Los péptidos son eluidos y separados por el uso de gradientes (Acetonitrilo (ACN) 3%, Ácido acético (HAc) 0,5 %, ACN 80% HAc 0,5%) para acceder directamente al espectrómetro de masas. Además, la elución es por flujo lento (100–200nl/min), y por tanto miles de péptidos pueden ser analizados mediante este método(52).
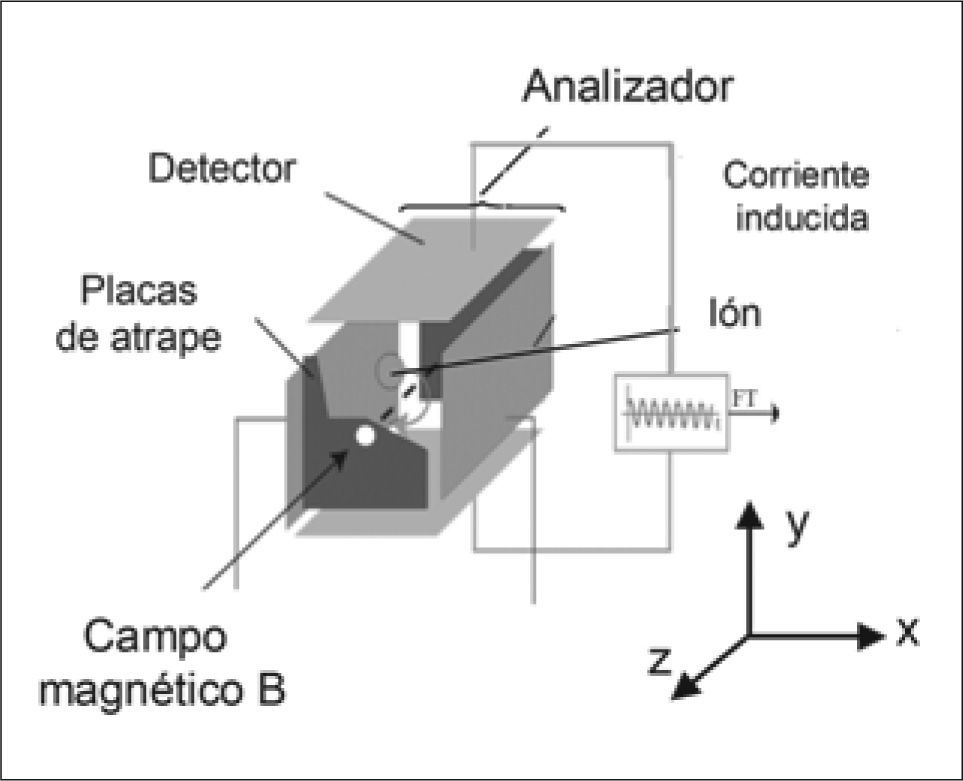
Utilidad de la disociación por captura electrónica ECD (Electrón Capture Dissociation) en un analizador FT-ICR MS (Fourier Transform Ion Cyclotron Resonance) para estudios de fosforilaciónEn 1974 el analizador de resonancia ciclotrónica de iones (ICR) fue descrito por Comisarow y Marshall(53). El principio general del instrumento FT-ICR se basa en dos fundamentos:
- a.
Los iones son atrapados en el interior de un campo magnético intenso. Si la velocidad de un ión es lenta y el campo magnético aplicado es intenso, el radio de la trayectoria del ión se hace pequeño. Entonces, el ión puede ser atrapado en una trayectoria circular que crea el campo magnético.
- b.
El campo magnético es orientado a lo largo del eje z y los iones son introducidos y atrapados a lo largo de este eje. En los planos x e y, los iones rotan alrededor del eje z. Cada ión lleva asociada una frecuencia que depende de su m/z. La medida de las frecuencias de las corrientes eléctricas inducidas permite obtener el valor de las masas de los iones. La señal es medida en el tiempo, siendo su intensidad proporcional al tiempo de medida.
En el analizador FT-ICR (Fig. 1), al crearse un sistema de vacío muy elevado, los iones generados en la fuente de ionización son guiados a través de una serie de bombas hasta que entran en una célula de movimiento circular (ICR). En dicha célula, la presión es de ~10-9Torr para que las trayectorias sean estables. Cuando los iones generados en la fuente de ionización entran en el analizador, éstos quedan expuestos a un campo magnético y a un campo eléctrico. El campo magnético fuerza a los iones a describir órbitas cerradas en un plano perpendicular a dicho campo, y las señales que se obtienen de las frecuencias ciclotrónicas de los iones, proporcionarán las masas de los analitos. El campo eléctrico permite variar el radio de las órbitas de los iones, para que éstos se posicionen cerca de las placas detectoras. Las señales de frecuencia que se obtienen serán transformadas a través de la FT para obtener los espectros (señal de intensidad en función de los valores m/z).
 se utiliza para analizar la señal producida, que es muy compleja. Figura adaptada de www.chm.bris.ac.ukyE.LopezVillar,Tesisdoctoral2006UCM-PRGroup.")
Esquema del analizador FT-ICR. El instrumento FT-ICR consiste en una trampa de iones que se encuentra en el interior de un imán que crea un campo magnético elevado y uniforme. Los iones son almacenados, analizados y detectados en el analizador y la transformada de Fourier (FT) se utiliza para analizar la señal producida, que es muy compleja. Figura adaptada de www.chm.bris.ac.ukyE.LopezVillar,Tesisdoctoral2006UCM-PRGroup.
Este instrumento suele ir acoplado a un sistema de adquisición de datos complejo, ya que al producir espectros de muy alta resolución, la cantidad de información producida es muy elevada(54).
El instrumento FT-ICR puede ser combinado también con la ionización en electrospray y técnicas de cromatografía líquida (HPLC), con lo que se obtiene una sensibilidad y precisión hasta ahora inigualable por otros espectrómetros de masas(55).
La fragmentación de péptidos ionizados vía ECD consiste en inducir la rotura del analito mediante disociación por captura de electrones. Esta fragmentación es alternativa a la fragmentación tipo CID, y típica de algunas trampas iónicas como la de Finnigan (LTQ; http://www.thermo.com/ com/cda/product/detail/1,,22172,00.htm).
McLafferty y cols.(56) introdujeron un método de fragmentación de péptidos/proteínas ionizadas: ECD. En este método, péptidos o proteínas multiprotonados son almacenados en un analizador trampa iónica con FT-ICR y expuestos a electrones con transferencia energética. La captura de un electrón por parte de un péptido protonado es exotérmica (6eV (1eV=1.602x10-19J) y causa que el péptido se fragmente.
La ECD en combinación con FT-ICR(41,42,45,53,54,57) permite que la fragmentación se produzca mayoritariamente en la cadena principal del péptido y escasamente en las cadenas laterales que incluyen PTMs. Esto es muy útil en estudios de fosforilación, y supone una ventaja frente a fragmentaciones con CID, donde es muy fácil perder el grupo fosfato (Fig. 2).
. Figura cedida por Karin Hjernø 2007 (PR Group).")
Diferentes técnicas de fragmentación en Espectrometría de Masas. CID: Disociación inducida por colisión. La molécula se fragmenta al adquirir un protón móvil. Éste tenderá a posicionarse en los aminoácidos básicos de la zona N-Terminal, inestabilizando la molécula y generándose las series de iones "b". Además, los péptidos trípticos normalmente tienen un residuo de lisina o arginina en el extremo C-terminal, así pues, ésta podrá también quedar secuestrada en el extremo C-terminal, que facilitaría la formación de la serie de iones "y".
ECD: Disociación por captura del electrón. La molécula se fragmenta al adquirir un electrón. Éste se unirá principalmente al C α del enlace amida de la cadena principal. Como consecuencia, la molécula se inestabiliza y se fragmenta, generándose finalmente mayoritariamente las series de iones iones "c" y "z". Preserva que la fragmentación se produzca en las cadenas laterales de la molécula, las cuales pueden contener modificaciones posttraduccionales (PTMs). Figura cedida por Karin Hjernø 2007 (PR Group).
Una desventaja del uso de ECD es la necesidad de que la muestra esté altamente limpia, junto con que su uso se desarrolla en espectrómetros de masas (ejemplo LTQ-FTICR) de muy elevado coste y requieren de personal especializado.
En un espectrómetro de masas LTQ-FT-ICR (Finnigan), el desarrollo de abordajes fosfoproteómicos consiste en que todos los iones entran en la trampa iónica lineal (LTQ), donde son almacenados y desde donde los iones son enviados a la FT-ICR para el análisis en modo MS con elevada resolución. Ahí, los iones de mayor intensidad (MS) son seleccionados para su fragmentación (MS/MS) en la trampa iónica lineal LTQ. Cuando se detectan iones con una pérdida neutral de ácido fosfórico (80-98Da), éstos serán de nuevo fragmentados (MS3) en la LTQ(51,52).
La ECD acoplada a FT-ICR, dada su elevada sensibilidad, es también un instrumento poderoso para la secuenciación de péptidos y proteínas.
CONCLUSIONES Y FUTURAS PERSPECTIVASActualmente, el campo de las rutas de señalización celulartan relevantes en patologías como el cáncer- está siendo bien equipado gracias a los avances e innovaciones de la proteómica y la espectrometría de masas. Las estrategias fosfoproteómicas y de MS actuales permiten identificar a gran escala residuos fosforilados de proteínas que componen diferentes cascadas de señalización. Por ello permiten profundizar en el entendimiento de las alteraciones en procesos de señalización y ayudan en el diseño y desarrollo de nuevas dianas terapeúticas(58).
Sin embargo, a pesar de los grandes avances previamente mencionados, a veces puede ser difícil disponer de suficiente cantidad de determinadas proteínas (por ejemplo algunas quinasas fosforiladas), lo cual dificulta en gran medida su análisis. Además, la preparación de la muestra a estudiar y los métodos a utilizar tienen que ser lo más óptimos y directos posible, con el fin de (a) minimizar pérdidas de las proteínas de interés, y (b) reproducir eficientemente los protocolos de la estrategia elegida.
Existen muchas y diferentes técnicas experimentales que permiten detectar la fosforilación (radioactividad, "western blotting", inmunoprecipitación con anticuerpos, etc) pero hasta hoy en día, la espectrometría de masas es la herramienta que permite identificar los residuos donde se produce dicha modificación post-traduccional. Esto último es de gran relevancia para el desarrollo farmacéutico de dianas específicas de drogas.
IMAC y Dióxido de titanio acoplados a espectrometría de masas, son una de las estrategias proteómicas de elección para la identificación de residuos fosforilados en serinas y treoninas.
Los anticuerpos anti-tirosina, han sido reciente y exitosamente utilizados para capturar proteínas fosforiladas en tirosina. Esta técnica, acoplada a espectrometría de masas, es una de las estrategias de elección para identificar residuos fosforilados en tirosinas.
La fragmentación ECD de péptidos ionizados, es una herramienta de gran utilidad en estudios de fosforilación. Su utilización implica una ventaja frente a fragmentaciones tipo CID, en las que es muy fácil perder el grupo fosfato. Esto último dificulta e incluso puede impedir la identificación del residuo fosforilado correspondiente a la o las proteínas a estudiar.
Finalmente, el gran y rápido avance de las metodologías proteómicas y de la espectrometría de masas en esta última década permite investigar diferentes patologías a nivel funcional. Para ello, siempre es necesario: (a) optimizar la preparación de la muestra (tejidos o líneas celulares), y (b) establecer estrategias proteómicas en función del tipo de muestra y de los objetivos científicos que se quieran alcanzar. Más aún, la elección del espectrómetro de masas a utilizar para el análisis de las muestras es también un requisito imprescindible que dependerá también de los objetivos finales del estudio a realizar.
AGRADECIMIENTOSLa Dra. E. López Villar disfruta de una beca de estancia postdoctoral "Marcos Fernández", Fundación Leucemia y Linfoma (F.L.L). Especial agradecimiento a Fernández Fermoselle, por su promoción de la investigación en cáncer, especialmente en leucemias y linfomas.
Agradecimientos también al Protein Research Group (PR Group, Department of Biochemistry and Molecular Biology, Odense University, Denmark) 2007, 2008; por la financiación del desarrollo experimental y su formación en el campo de la Proteómica y la Espectrometría de Masas en Odense. Especiales agradecimientos al Prof. Dr. Ole Nørregaard Jensen y al Prof. Dr. Peter Roepstorff, así como a la Dra. Julia Sequí, por sus directrices en el campo de la inmunología.
Las figuras adjuntadas en esta revisión han sido cedidas por el "Protein Research Group", Odense, y se detallan también en la Tesis Doctoral "Abordajes Proteómicos para el estudio de la Secreción No Clásica y Fosforilación en Saccharomyces cerevisiae" E. López Villar, Octubre 2006 (UCM, España, en colaboración con PR Group, Dinamarca).
CONFLICTO DE INTERÉSLos autores declaran no tener conflicto de interés.