


Se incluyen las aportaciones restantes de la VIII edición de las reuniones del Grupo Español para las Inmunodeficiencias Primarias (IDP), celebradas en Octubre de 2007, en las que han participado más de ochenta especialistas en diversos campos (Inmunología, Pediatría, Alergia, Inmunología Clínica) directamente implicados en esta problemática.
We present the pending contributions to the VIII edition of the Meeting of the Spanish Group for Primary Immunodeficiencies (PI). More than eighty specialists belonging to different Spanish groups interested in the study of immunodeficiency from different points of view, including immunologists, pediatricians, allergologists or clinical immunologists, attended the meeting, held in October 2007.
J. de Gracia, A. Álvarez
Servicio de Neumología, Hospital Vall d´ Hebron, Barcelona.
Los mecanismos de defensa en las vías respiratorias son múltiples, y van desde elementos físicos como el sistema mucociliar a la participación de las defensas locales y la función de los anticuerpos y las células de la respuesta inmunológica.
La IgA predomina en las vías aéreas superiores y la IgG y sus subclases en las vías aéreas inferiores. Estas inmunoglobulinas o anticuerpos impiden la colonización de estas vías por los gérmenes que entran del exterior.
Los linfocitos participan en la respuesta inmunitaria local cuando se produce una agresión bacteriana o viral y, después de la reacción inflamatoria inicial, se activan para dar lugar a las células productoras de anticuerpos y células citotóxicas.
Las principales manifestaciones clínicas, cuando la respuesta inmunológica no es adecuada, son las infecciones de las vías aéreas superiores (p.e., sinusitis y otitis) e inferiores como las neumonías(1) producidas por bacterias, gérmenes encapsulados y mayor incidencia de Mycoplasma neumoniae.
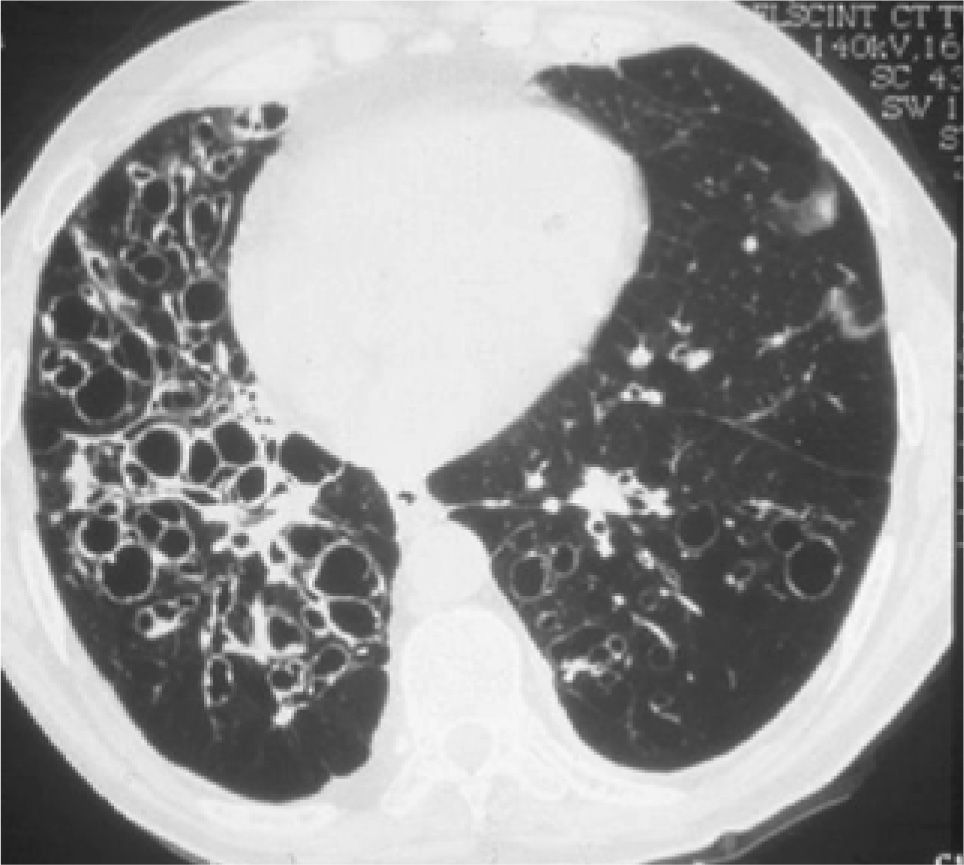
Las infecciones crónicas dan lugar a las bronquiectasias (Fig. 1) y la insuficiencia respiratoria, que son las causas más frecuentes de morbi-mortalidad en estos pacientes(2-4).

El tratamiento substitutivo de los defectos de la producción de anticuerpos, como son la Inmunodeficiencia Variable Común (IDVC), la Agammaglobulinemia ligada al cromosoma X, los defectos de producción de anticuerpos, etc., con gammaglobulinas (GG) por vía endovenosa (e.v.) o subcutánea (s.c.) reduce tanto la incidencia de infecciones como el daño pulmonar(5), y mejora el pronóstico de los pacientes. Para ello es necesario mantener los niveles de IgG en suero por encima de 600 mg/100 ml, antes de una nueva administración.
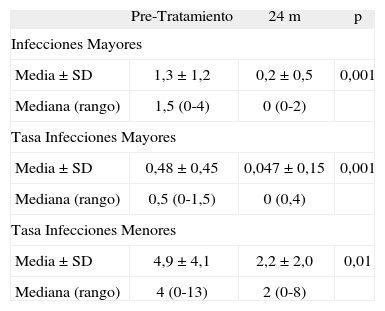
En un estudio realizado en nuestro hospital con pacientes en tratamiento con gammaglobulina endovenosa, se han conseguido los resultados que se muestran en la Tabla I.
Infecciones en pacientes con IDVC antes y después del tratamiento con GG e.v.
| Pre-Tratamiento | 24 m | p | |
| Infecciones Mayores | |||
| Media ±SD | 1,3 ± 1,2 | 0,2 ± 0,5 | 0,001 |
| Mediana (rango) | 1,5 (0-4) | 0 (0-2) | |
| Tasa Infecciones Mayores | |||
| Media ± SD | 0,48 ± 0,45 | 0,047 ±0,15 | 0,001 |
| Mediana (rango) | 0,5 (0-1,5) | 0 (0,4) | |
| Tasa Infecciones Menores | |||
| Media ± SD | 4,9 ± 4,1 | 2,2 ± 2,0 | 0,01 |
| Mediana (rango) | 4 (0-13) | 2 (0-8) | |
Estos pacientes precisan, además del tratamiento substitutivo con GG's(6,7), medidas de soporte como la fisioterapia, anti-inflamatorios, broncodilatadores y antibióticos, según las infecciones detectadas. Las medidas terapéuticas actuales, junto con un diagnóstico precoz del defecto inmunológico, han mejorado la calidad de vida y el pronóstico de estos enfermos.
NUESTRA EXPERIENCIA EN EL DIAGNÓSTICO Y TRATAMIENTO DE LA ENFERMEDAD GRANULAMATOSA CRÓNICAIsabel Caragol
Unidad de Inmunología. Hospital Universitario Vall d'Hebron, Barcelona.
La enfermedad granulomatosa crónica (EGC) es una inmunodeficiencia primaria que afecta la función bactericida de los fagocitos, causada por mutaciones en cualquiera de los componentes del sistema oxidativo NADPH oxidasa, gp91, p47, p67 y p22(1). La incidencia de esta enfermedad se estima que es de 1:250,000, y su diagnóstico se basa en la sospecha clínica y el estudio de la capacidad oxidativa de los granulocitos. La mayoría de los casos se diagnostican en la edad pediátrica, antes de los 3 años de edad, pero por desconocimiento médico de la enfermedad, hay pacientes que llegan a la edad adulta sin diagnosticar. Las infecciones que padecen estos enfermos son características y de extrema gravedad en muchos casos. Es por lo tanto una enfermedad de mortalidad elevada. El tratamiento se basa en la antibioticoterapia precoz y agresiva de las infecciones y una profilaxis frente a infecciones bacterianas y fúngicas, que, si bien reduce considerablemente el número de infecciones, no las previene totalmente, siendo cada vez más clara la indicación de trasplante de precursores hematopoyéticos, la única alternativa actual de curación(2).
Es por esta razón que todos los esfuerzos en conocer y dar a conocer esta enfermedad, las manifestaciones clínicas más frecuentes, y todo lo que ayude al diagnóstico precoz de la misma son de extrema relevancia como primer paso para reducir su mortalidad y morbilidad.
Hemos revisado los casos diagnosticados en nuestro centro desde 1980 hasta la actualidad, recogiendo, entre otros datos, la clínica y edad del debut, los gérmenes implicados en las infecciones, la edad al diagnóstico, el tratamiento profiláctico, la evolución, mortalidad, diagnóstico inmunológico y genético y la evolución a lo largo de este periodo de 27 años.
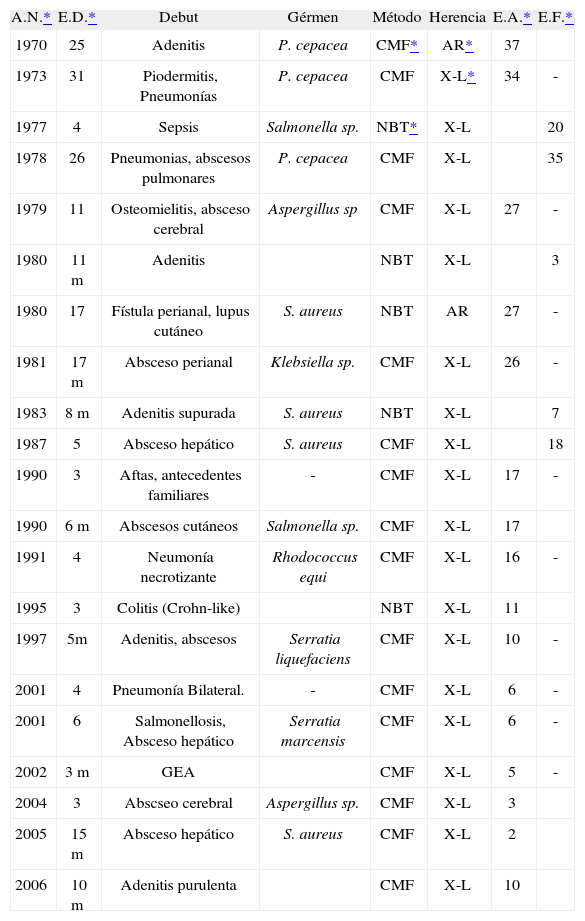
En la Tabla I se expresan los datos individualizados de los pacientes. En este periodo se han diagnosticado un total de 21 casos, 18 en la edad pediátrica y 3 en la edad adulta. La mediana de edad al diagnóstico de los 18 niños fue de 27 meses (3 meses-17 años), mientras que la edad media al diagnóstico de los 3 adultos fue de 27 años.
Datos clínicos e inmunológicos de los pacientes con EGC
| A.N.* | E.D.* | Debut | Gérmen | Método | Herencia | E.A.* | E.F.* |
| 1970 | 25 | Adenitis | P. cepacea | CMF* | AR* | 37 | |
| 1973 | 31 | Piodermitis, Pneumonías | P. cepacea | CMF | X-L* | 34 | - |
| 1977 | 4 | Sepsis | Salmonella sp. | NBT* | X-L | 20 | |
| 1978 | 26 | Pneumonias, abscesos pulmonares | P. cepacea | CMF | X-L | 35 | |
| 1979 | 11 | Osteomielitis, absceso cerebral | Aspergillus sp | CMF | X-L | 27 | - |
| 1980 | 11 m | Adenitis | NBT | X-L | 3 | ||
| 1980 | 17 | Fístula perianal, lupus cutáneo | S. aureus | NBT | AR | 27 | - |
| 1981 | 17 m | Absceso perianal | Klebsiella sp. | CMF | X-L | 26 | - |
| 1983 | 8 m | Adenitis supurada | S. aureus | NBT | X-L | 7 | |
| 1987 | 5 | Absceso hepático | S. aureus | CMF | X-L | 18 | |
| 1990 | 3 | Aftas, antecedentes familiares | - | CMF | X-L | 17 | - |
| 1990 | 6 m | Abscesos cutáneos | Salmonella sp. | CMF | X-L | 17 | |
| 1991 | 4 | Neumonía necrotizante | Rhodococcus equi | CMF | X-L | 16 | - |
| 1995 | 3 | Colitis (Crohn-like) | NBT | X-L | 11 | ||
| 1997 | 5m | Adenitis, abscesos | Serratia liquefaciens | CMF | X-L | 10 | - |
| 2001 | 4 | Pneumonía Bilateral. | - | CMF | X-L | 6 | - |
| 2001 | 6 | Salmonellosis, Absceso hepático | Serratia marcensis | CMF | X-L | 6 | - |
| 2002 | 3 m | GEA | CMF | X-L | 5 | - | |
| 2004 | 3 | Abscseo cerebral | Aspergillus sp. | CMF | X-L | 3 | |
| 2005 | 15 m | Absceso hepático | S. aureus | CMF | X-L | 2 | |
| 2006 | 10 m | Adenitis purulenta | CMF | X-L | 10 |
Las manifestaciones clínicas al debut, así como los gérmenes aislados, no difirieron entre los dos grupos de edad, siendo las más frecuentes por este orden los abscesos, las adenitis supuradas o abscesificadas y las neumonías, sumando entre las tres más del 60% del total de 10 diferentes manifestaciones recogidas como: osteomielitis, sepsis, GEA, colitis, piodermitis, y otras. Los gérmenes aislados más frecuentemente, como responsables de las infecciones, fueron por este orden: Stafilococcus aureus, Salmonella sp., Pseudomona cepacea, Aspergillus sp. y Klebsiella sp.
En 19 de los 21 casos la forma de EGC fue ligada al cromosoma X, y por lo tanto eran varones con una mutación en la proteína gp91 cuya madre presentaba el típico patrón de portadora con dos poblaciones de granulocitos, excepto en un caso en que fue una mutación de novo. Los dos casos de herencia autosómica recesiva fueron mujeres con mutaciones en la p67, una de ellas diagnosticada a los 17 años, catalogada como portadora sintomática desde los 13 años y la otra a los 25 años. El diagnóstico inmunológico se hizo mediante el test de reducción del nitroazul de tetrazolio (NBT) en cinco casos. A partir de 1993 se empezó a utilizar la citometría de flujo junto con la dihidrorodamina (DHR 123)(3), un método mucho más sensible y objetivo, mediante el cual se realizó el diagnóstico en los 16 casos restantes. El estudio genético se realizó en 17 (71%) de los casos y en distintos centros: Zurich (JP Hossle), Amsterdam (D Roos) y Madrid (A Ferreira). En el resto de los casos, 2 fallecieron sin estudio y 3 están en estudio actualmente.
La mortalidad de esta serie es del 23,4% (5 casos), que fallecieron a una edad media de 12 años. Las causas del fallecimiento fueron por complicaciones de un trasplante renal, neumopatía infecciosa, sepsis por germen Gram-, aspergillosis, y síndrome hemofagocítico secundario a leishmaniosis. Llama la atención que en esta serie hubo otro caso con infección por Leishmania, con síndrome hemofagocítico asociado, que evolucionó favorablemente.
Los pacientes recibieron cotrimoxazol (89%), itraconazol (79%) e interferón gamma recombinante (42%).
Si calculamos el número de casos de EGC diagnosticados por década, 1981-1990, 1991-2000 y 2001-2007, observamos que este ha ido en aumento (0,4, 0,9 y 1,1 casos/año, respectivamente), aún mas teniendo en cuenta que el tercer periodo es de 7 años. Estos datos podrían indicar un progreso en el conocimiento y la conciencia de la existencia de esta enfermedad.
En esta serie de pacientes destaca como poco frecuente, según la literatura, la incidencia de infección por Leishmania(4) así como la mutación en la p67 en los dos casos de herencia autosómica recesiva. Respecto al diagnóstico, queremos insistir en la importancia de la sospecha clínica y la validez de la citometría de flujo como método de elección para pacientes y familiares portadoras. El tratamiento que actualmente ofrece más expectativas es el trasplante de precursores hematopoyéticos, en espera de que los resultados de la investigación sobre terapia génica sean más concluyentes.
EXPERIENCIA EN EL TRANSPLANTE DE PRECURSORES HEMATOPOYÉTICOS EN INMUNODEFICIENCIASTeresa Olivé
Servicio de Oncohematología. Hospital Infantil Vall d'Hebron, Barcelona.
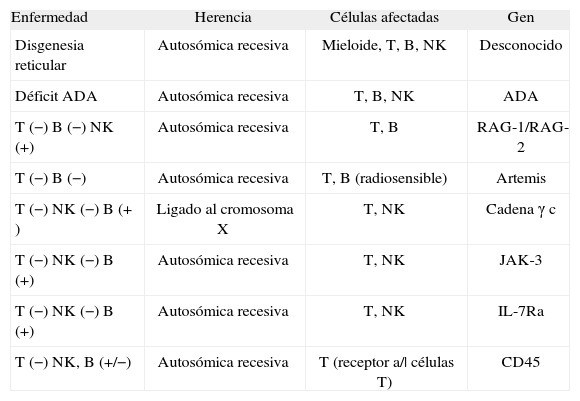
INTRODUCCIÓNEn las Inmunodeficiencias Severas Combinadas (IDSC) se incluyen un grupo de enfermedades que se caracterizan por un bloqueo en el desarrollo de los linfocitos T, asociado a una alteración de otras líneas linfocitarias (B, NK o más raramente línea mieloide)(1). Se distinguen 9 enfermedades en función del fenotipo y patrón de herencia (Tabla I). Su frecuencia global estimada es de 1/75.000-100.000 nacimientos. La sintomatología clínica se inicia muy precozmente en forma de infecciones respiratorias e intestinales, candidiasis oral, diarrea persistente, provocando un estacionamiento pondoestatural, malnutrición y neumonías intersticiales debidas a gérmenes oportunistas (Pneumocystis jirovecii, Aspergillus, organismos intracelulares, virus del grupo herpes)(2). Otras manifestaciones no infecciosas incluyen la posibilidad de presentar una enfermedad injerto contra el huésped (EICH) ya sea a partir de linfocitos de origen materno, o por la transfusión de hemoderivados. El implante de linfocitos maternos se presenta en un 50% de IDSC, y no se observa en los pacientes con déficit de adenosín deaminasa (ADA). La manifestación clínica más importante es el exantema cutáneo con infiltración linfoide junto con eosinofilia, afectación hepática, con aumento de enzimas hepáticos, e infiltrado linfoide periportal. Dicha alteración es controlable y no tiene efectos letales. Por el contrario, la inoculación postnatal de linfocitos a través de hemoderivados puede dar lugar a una EICH grave, generalmente de consecuencias letales(3).
Inmunodeficiencias graves combinadas
| Enfermedad | Herencia | Células afectadas | Gen |
| Disgenesia reticular | Autosómica recesiva | Mieloide, T, B, NK | Desconocido |
| Déficit ADA | Autosómica recesiva | T, B, NK | ADA |
| T (−) B (−) NK (+) | Autosómica recesiva | T, B | RAG-1/RAG-2 |
| T (−) B (−) | Autosómica recesiva | T, B (radiosensible) | Artemis |
| T (−) NK (−) B (+ ) | Ligado al cromosoma X | T, NK | Cadena γ c |
| T (−) NK (−) B (+) | Autosómica recesiva | T, NK | JAK-3 |
| T (−) NK (−) B (+) | Autosómica recesiva | T, NK | IL-7Ra |
| T (−) NK, B (+/−) | Autosómica recesiva | T (receptor a/| células T) | CD45 |
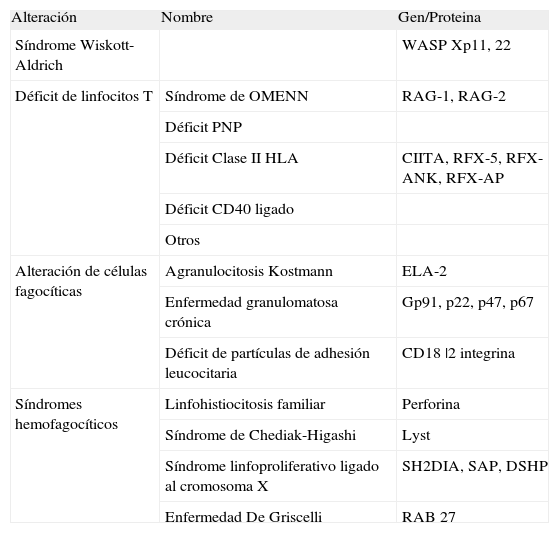
Otras immunodeficiencias no IDSC incluyen las siguientes: Síndrome de Wiskott-Aldrich, trastornos de la fagocitosis (déficit de partículas de adhesión leucocitaria y enfermedad granulomatosa crónica) y síndromes hemofagocíticos (linfohistiocitosis familiar, síndrome de Chediak-Higashi, enfermedad de Griscelli, síndrome linfoproliferativo ligado al cromosoma X) (Tabla II).
Otras inmunodeficiencias que son tratadas con TPH
| Alteración | Nombre | Gen/Proteina |
| Síndrome Wiskott-Aldrich | WASP Xp11, 22 | |
| Déficit de linfocitos T | Síndrome de OMENN | RAG-1, RAG-2 |
| Déficit PNP | ||
| Déficit Clase II HLA | CIITA, RFX-5, RFX-ANK, RFX-AP | |
| Déficit CD40 ligado | ||
| Otros | ||
| Alteración de células fagocíticas | Agranulocitosis Kostmann | ELA-2 |
| Enfermedad granulomatosa crónica | Gp91, p22, p47, p67 | |
| Déficit de partículas de adhesión leucocitaria | CD18 |2 integrina | |
| Síndromes hemofagocíticos | Linfohistiocitosis familiar | Perforina |
| Síndrome de Chediak-Higashi | Lyst | |
| Síndrome linfoproliferativo ligado al cromosoma X | SH2DIA, SAP, DSHP | |
| Enfermedad De Griscelli | RAB 27 |
La evolución natural de estas enfermedades es mala, y la mayoría de los pacientes fallecen antes del año de vida. Una vez efectuado el diagnóstico, y a la espera del tratamiento definitivo que consiste en un transplante de precursores hematopoyéticos (TPH), deben iniciarse medidas de aislamiento y profilaxis de las infecciones con: cotrimoxazol, antifúngicos, antivíricos, administración periódica de inmunoglobulinas, e irradiación de los hemoderivados. Solo en los pacientes con déficit de ADA, se efectuará tratamiento sustitutivo con PEG-ADA semanal, con efectividad en el 90%(4).
El TPH a partir de un hermano HLA idéntico es el tratamiento de elección conocido ya desde hace más de 30 años, y el único con finalidades curativas. Sin embargo, no todos los pacientes afectos de inmunodeficiencias disponen de un hermano idéntico, por lo que se han investigado fuentes de obtención y tipos de donantes alternativos (haploidéntico, no familiar).
Los pacientes con IDSC con donante genotípicamente idéntico no requieren tratamiento de condicionamiento ni profilaxis de la EICH. En cambio cuando se trata de un donante no familiar o familiar fenotípicamente idéntico se tratan con Busulfan+Ciclofosfamida, asociado o no a ATG y Ciclosporina A. En los donantes haploidénticos se efectúa una depleción linfoide T. En el resto de immunodeficiencias no IDSC sí que se efectúa tratamiento de acondicionamiento, combinando Busulfan + Ciclofosfamida + ATG.
Recientemente se han publicado los resultados del TPH en inmunodeficiencias en Europa a partir del registro SCETIDE (European Group for Blood and Marrow Transplantation and the European Society for Immunodeficiency) (5). Se ha analizado la evolución de 919 pacientes afectos de IDSC y otras inmunodeficiencias no severas combinadas, transplantados entre 1968 y 1999 en 37 centros correspondientes a 18 países. Esto suma un total de 1072 TPH en 919 pacientes (566 TPH en 475 IDSC, y 512 TPH en 444 no IDSC). La mediana de seguimiento es de 9 años en IDSC y de 7 años para no IDSC. La fuente de obtención más frecuente fue la médula ósea (87.3%), seguida de la sangre periférica (12%) y la sangre de cordón umbilical (0,7%). 205 pacientes con IDSC no recibieron tratamiento de condicionamiento, y el resto lo recibieron de acuerdo con las recomendaciones del "European Group for Blood and Marrow Transplantation and European Society for Immunodeficiency Working Group". Excepto en dos casos, todos los pacientes con otras inmunodeficiencias, recibieron Busulfan (16-20 mg/kg) y Ciclofosfamida (200 mg/kg). La depleción linfoide T se efectuó en el 91% de los donantes haploidénticos y en el 41% de los no familiares. La profilaxis de la EICH se efectuó con Methotrexato antes del 1983, y posteriormente con Ciclosporina A y Methotrexato pauta corta.
La supervivencia a 3 años en los pacientes con IDSC fue superior en los HLA idénticos en comparación con los haploidénticos (77% frente a 54%; p=0.002). No existieron diferencias en el grupo de donantes HLA idénticos [genotípico, fenotípico familiar y no familiar (81%, 72%, y 63%)].
En los pacientes con otras inmunodeficiencias no severas combinadas la supervivencia también fue superior en los donantes HLA idénticos comparados con los haploidénticos [genotípico HLA idéntico 71%; fenotípico HLA idéntico 42%; haploidéntico 42%; no familiar 59% (p= 0,0006)].
En el análisis de 919 pacientes afectos de immunodeficiencias a partir del "International Bone Marrow Transplant Registry" (IBMTR) pertenecientes a 111 grupos, desde 1980 a 1998 se incluyen 428 IDSC, 50 síndrome de Omenn; 223 síndrome de Wiskott-Aldrich, 34 síndrome Chediak-Higashi, 57 síndrome linfoproliferativo ligado al cromosoma X; 23 síndrome de Kosttman, y 104 inmunodeficiencias combinadas. En el 53% de los casos el donante fue un familiar alternativo, en el 27% un hermano idéntico, y en el 20% un donante no familiar. La fuente de obtención fue mayoritariamente a partir de médula ósea (93%). La cuarta parte de los pacientes no recibió tratamiento de condicionamiento, y en el resto, más de la mitad recibieron Busulfan + Ciclofosfamida. En IDSC la probabilidad de supervivencia fue del 84% empleando como donante a un hermano idéntico, del 65% con donante no familiar y del 55% con familiar alternativo. En otras inmunodeficiencias dicha probabilidad a partir de un hermano idéntico, no familiar y familiar alternativo fue: síndrome de Omenn, 65%, 63% y 45%; síndrome de Wiskott-Aldrich, 90%, 68% y 60%; síndrome de Chediak-Higashi, 80%, 68% y14%, respectivamente.
El grupo del Duke University Medical Center, publicó en 1999 los resultados de un total de 89 pacientes trasplantados en un único centro entre mayo de 1982 y septiembre de 1998(6). De éstos, 77 recibieron médula ósea haploidéntica deplecionada, de los cuales tres recibieron además un trasplante de sangre de cordón umbilical y otros doce, médula ósea de un familiar idéntico. Ninguno recibió tratamiento de acondicionamiento ni profilaxis de la EICH, con la excepción de dos pacientes que recibieron sangre de cordón umbilical. El 81% de los pacientes viven, con una mediana de seguimiento de 5 años y medio. La recuperación de la función linfoide T fue total en los 72 supervivientes, excepto en 4. En 45 de 72, se siguió un tratamiento con inmunoglobulinas intravenosas.
En un estudio cooperativo de IBMTR y NMDP(7) sobre 170 pacientes con síndrome de Wiskott-Aldrich y trasplantados entre 1968 y 1996, la probabilidad de supervivencia a partir de familiar idéntico, familiar alternativo y no familiar fue de 87%, 52% y 71% respectivamente.
En el hospital Vall d'Hebron de Barcelona, 49 pacientes fueron sometidos a un TPH entre febrero de 1995 y junio de 2007. 24 de ellos eran afectos de IDSC y 25 no IDSC (8 enfermedad de Wiskott-Aldrich, 6 Linfohistiocitosis hemofagocítica familiar, 2 Síndrome de Omenn, 3 Agranulocitosis de Kostman, 2 Síndrome linfoproliferativo ligado al cromosoma X, 1 déficit de partículas de adhesión leucocitaria, 1 enfermedad de Chediak Higashi, 1 déficit de clase II HLA y 1 déficit de CD40 ligando). Según el tipo de donante, 12 de un hermano HLA idéntico, 13 de un donante haploidéntico y 24 de un donante no familiar. La fuente de obtención fue la médula ósea (23 casos), sangre periférica (8 casos) y sangre de cordón umbilical (18 casos). En 44 de 49 pacientes se efectuó tratamiento de acondicionamiento. La profilaxis de la EICH consistió en Ciclosporina A, más/menos Methotrexato asociado a Globulina Antitimocítica (GAT) y depleción linfoide T en aquellos pacientes con TPH haploidéntico.
RESULTADOSSe observa fallo de implante primario en 2 pacientes: uno afecto de disgenesia reticular y otro con un déficit de CD18. Fallo de implante secundario en 3 pacientes: 1 de enfermedad de Wiskott-Aldrich y 2 de linfohistiocitosis hemofagocítica familiar.
EVOLUCIÓNSupervivencia 30/49 (61,2%) con una mediana de seguimiento de 7 años. Dependiendo del tipo de donante: 91% de hermano idéntico; 30% haploidéntico, y no familiar, 62%. Según la fuente de obtención: médula ósea, 60%; sangre periférica, 37% y sangre de cordón umbilical, 72%. No existen diferencias en cuanto al tipo de inmunodeficiencia. Evolución clínica de los pacientes vivos: en 27 la recuperación inmunológica fue total; 5 presentaron fenómenos de autoinmunidad, 1 fibrosis pulmonar agresiva, 1 hipertensión pulmonar de causa no filiada que requirió un trasplante pulmonar, presentando posteriormente un síndrome linfoproliferativo. De los pacientes fallecidos (19 de 49, 39,8%), en 6 la causa de muerte fue la progresión de la enfermedad, en 6 una EICH aguda III-IV, en 3 neumopatía progresiva, en 1 encefalitis subaguda, en 1 síndrome proliferativo, en 1 hemorragia cerebral y en 1 una cirrosis hepática secundaria a hepatitis C.
CONCLUSIONES- 1.
El TPH ha sido y es el tratamiento de elección de un determinado grupo de inmunodeficiencias desde hace más de 30 años.
- 2.
En ausencia de un donante familiar idéntico, un TPH haploidéntico con depleción linfoide T era una indicación en la época de los 80.
- 3.
La instauración de registros de donantes voluntarios de precursores hematopoyéticos, junto con la existencia de bancos de sangre de cordón umbilical han permitido mejorar los resultados del TPH en inmunodeficiencias.
- 4.
La probabilidad de supervivencia en todas las series es superior con donantes idénticos (familiar y no familiar) en comparación con los no idénticos.