


La Ataxia Telangiectasia (AT) es un síndrome multisistémico complejo que se hereda de forma autosómica recesiva y está incluida dentro de los síndromes genéticos de inestabilidad cromosómica. Las mutaciones en el gen ATM (ataxia telangiectasia mutado) son la causa de la aparición de la AT. La susceptibilidad a las infecciones recurrentes o persistentes de severidad variable es una característica principal en el 70% de los individuos con AT y son debidos en parte a la inmunodeficiencia observada en estos pacientes. La AT es un trastorno muy raro incluido en los síndromes de Inmunodeficiencia Primaria. Este ocurre con una frecuencia de 1 caso por 40 a 100 000 nacidos en el mundo, con prevalencia de portadores de 1.4–2%. Se presenta un paciente masculino de 10 años de edad, en el que a los 2 años de edad, se constató ataxia de tronco y de la marcha, hipotonía muscular generalizada y disartria. El paciente también desarrolló en piel y mucosas hiperpigmentación e hipopigmentación. A los 3 años de edad aparecieron telangiectasias oculocutáneas y se detectaron nistagmos a los 6 años y 10 meses de edad. Por otra parte, comenzó a presentar episodios frecuentes de infecciones respiratorias altas y bajas: rinitis purulenta, sinusitis maxilar, y bronconeumonías tan bien como giardiasis. Los estudios inmunológicos a los 2 años de edad revelaron una disminución de las concentraciones séricas de IgG, IgA, e IgM aumentada; niveles bajos de IgE y elevados de ·-fetoproteína. La RMN a los 7 años de edad mostró una atrofia cerebelosa. Los linfocitos T CD2+ CD8+ estaban disminuidos. El paciente fue tratado con gammaglobulina, dieta balanceada y vitaminoterapia con evolución satisfactoria a pesar del pronóstico reservado de este síndrome que se caracteriza por neurodegeneración progresiva.
Ataxia telangiectasia (AT) is a multisystemic complex syndrome that is typically transmitted in an autosomal recessive mode of inheritance and it is included among chromosomal breakage syndromes. The mutations on Ataxia-telangiectasia mutated (ATM) gen are the cause of AT disease. The risk to recurrent infection, with variable severity, is observed in 70 percent of these patients and it is attributed to their immunodeficiency. AT is a very rare disorder included in the Primary Immunodeficiency Syndromes. It occurs at a frequency of 1 case per 40 to 100,000 births worldwide with an estimated frequency of carriers of 1.4–2%. We present a case of a ten year- old male who had a past personal history of truncal and walking ataxia, generalized muscular hypotonia, and dysarthria beginning at age two. He also developed hyper and hypopigmentation in the skin and mucosa. At the age of three, an oculo-cutaneous telangiectasia appeared and nystagmus was found when he was six years and ten months old. On the other hand, he also began to suffer frequent episodes of infections of the upper and the lower respiratory tracts including: purulent rhinitis, maxilar sinusitis and bronchopneumonias as well as giardhiasis. Immunologic studies at the age of two revealed a decrease of the seric concentrations of IgG, IgA, high IgM values; low levels of IgE and high levels of alpha-fetoprotein. At seven, the MRI showed cerebellar atrophy. The amount of CD2+ CD8+ T lymphocytes was diminished. The patient was treated with gammaglobulin, balanced diet and vitamin therapy with satisfactory evolution in spite of the reserved prognosis for this syndrome that is characterized by a progressive neurodegeneration.
La Ataxia-Telangiectasia (A-T), síndrome de BoderSedgwick o síndrome de Louis-Bar es un trastorno neurodegenerativo hereditario autosómico recesivo multisistémico complejo raro, que afecta a una variedad de órganos en el cuerpo y causa discapacidad severa, se presenta tempranamente en la niñez y no tiene predilección por sexo. La ataxia cerebelar temprana de desarrollo progresivo significa dificultad con el control del movimiento (marcha inestable), y las telangiectasias son vasos sanguíneos extras, que pueden ser vistos particularmente en la conjuntiva bulbar de los ojos. La enfermedad es heterogénea, clínica y genéticamente, lo que se muestra por la existencia de 4 grupos de complementación (A, C, D, E)(1-6).
Syllaba y Henner publicaron por primera vez las descripciones de pacientes con A-T en 1926(7). Ellos observaron coreoatetosis progresiva y telangiectasia ocular en 3 miembros de una única familia. En 1941, Louis-Bar, describió ataxia cerebelar progresiva y telangiectasia cutánea en un niño belga(8). El síndrome recibió el nombre de Louis-Bar. La AT no fue descrita como una entidad clínica distintiva hasta que Boder, Sedgwick y Biemond en 1957, con la ayuda de autopsias, reportaron anormalidades del desarrollo de órganos, manifestaciones neurológicas, y una tercera característica importante de la enfermedad: las infecciones senopulmonares recurrentes(9,10).
A-T es progresiva y afecta el cerebelo (el centro del control motor del cuerpo) y, en cerca del 70% de los casos, también se afecta el sistema inmune, conduciendo a trastornos respiratorios(11-14).
La A-T es reportada en todas las regiones del mundo. La incidencia probable es aproximadamente de 1 caso en 40 a 100,000 nacidos(15) y la frecuencia de la heterocigocidad del alelo mutante de A-T fue reportada ser de 1.4-2% de la población general(15,16).
El diagnóstico de la Ataxia-Telangiectasia se basa en los hallazgos clínicos característicos y los resultados de los análisis clínicos. El período en el que es más difícil diagnosticar la A-T es en la infancia temprana cuando aparecen por primera vez los síntomas neurológicos y en el que las telangiectasias típicas aún no han aparecido. Durante este período, un historial de infecciones recurrentes y hallazgos inmunológicos típicos pueden ser sugerentes del diagnóstico(17-19). Una de las pruebas de laboratorio de más ayuda que se utiliza para asistir en el diagnóstico de A-T es la medida de proteínas fetales en la sangre, que pueden persistir en altos niveles en sangre en algunas afecciones (tales como A-T) después del nacimiento. La gran mayoría de los pacientes con A-T (>95%) presentan niveles elevados de α-fetoproteína en suero. Cuando se han eliminado otras causas de elevación de α-fetoproteína, la α-fetoproteína elevada en sangre, en asociación con los signos y síntomas característicos, hacen el diagnóstico de A-T casi certero(20). Los individuos con AT pueden también tener una mayor frecuencia de roturas espontáneas en sus cromosomas, así como una mayor frecuencia de reacomodos de cromosomas. Estas anormalidades ocurren frecuentemente en la cercanía de genes esenciales para la función de linfocitos, tales como los genes receptores de antígenos de inmunoglobulina y linfocitos T. La frecuencia de roturas de cromosomas aumenta cuando los linfocitos T son expuestos a rayos X en el laboratorio, y esto conforma las bases para una prueba de diagnóstico especializada para la A-T(21-24).
El gen responsable de la A-T (gen ATM; gen mutado de la ataxia-telangiectasia) ha sido identificado y se encuentra en el brazo largo del cromosoma 11 en 11q22-23(25,26). Éste controla la producción de una enzima del tipo fosfatidilinositol- 3-cinasa involucrada en el control del ciclo celular, la recombinación de ADN, la apoptosis y otras respuestas celulares al daño de ADN. Las anormalidades en este control mediado por ATM producen ADN dañado, con lo cual se acumulan las roturas cromosómicas con el paso del tiempo y la célula muere. Estos fenómenos afectarían especialmente los timocitos, los linfocitos B inmaduros, las células de Purkinje del sistema nervioso central y el endotelio vascular(27,28). La identificación del gen específico responsable de la A-Tha hecho posible la detección de portadores y el diagnóstico prenatal, sin embargo aún no se encuentra disponible en laboratorios comerciales.
No existe cura para ninguno de los problemas de la Ataxia-Telangiectasia, y aunque no hay tratamiento específico disponible, es de sostén en gran medida, se investigan estrategias terapéuticas potenciales(29,30). Se debe promover la participación de los pacientes en tantas actividades como sea posible, y deben recibir apoyo para mantener un estilo de vida tan normal como sea posible.
La evolución de la enfermedad en la mayoría de los pacientes se caracteriza por el deterioro neurológico progresivo. Muchos pacientes son confinados a una silla de ruedas en su adolescencia. Las infecciones en pulmón (bronquitis o neumonía) y senos paranasales (sinusitis) son comunes y pueden dañar los pulmones aún cuando sean tratadas a tiempo. Las neoplasias o cáncer son también más comunes en los pacientes con A-T. Pueden ser tratadas pero pueden requerir modificaciones de los protocolos estándares de quimioterapia. Sin embargo, aún cuando el curso que se menciona es el más típico, el curso de la A-T varía considerablemente de paciente a paciente. Algunos pacientes han podido asistir a la universidad, y algunos han vivido hasta la quinta década de vida(31,32).
En este trabajo se presenta el caso de un niño con AtaxiaTelangiectasia, una inmunodeficiencia primaria.
MATERIALES Y METODOSPreviamente a la realización del reporte del caso se obtuvo el consentimiento por escrito de la madre del paciente, quien estuvo de acuerdo en la realización de estudios inmunológicos con toma de muestras de sangre, prueba cutánea, estudios radiológicos y la obtención de fotos del paciente. Se incluyó al paciente con el diagnóstico de Ataxia Telangiectasia a los 2 años de edad, que se siguió evolutivamente por la consulta de inmunología hasta los 10 años.
Todas las determinaciones fueron realizadas a ciegas en el laboratorio clínico.
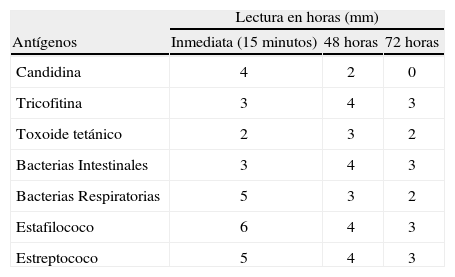
Se determinó la cuantificación de inmunoglobulinas(33). Se estudió la inmunidad celular mediante la determinación de subpoblaciones linfocitarias por citometría de flujo (34), y por la aplicación de la prueba cutánea de hipersensibilidad retardada(35) en el paciente utilizando los siguientes extractos de cepas bacterianas y micóticas: toxoide tetánico, bacterias intestinales, bacterias respiratorias, estafilococo, estreptococo, candidina y tricofitina.
CASO CLÍNICO Y DISCUSIONLa Ataxia-Telangiectasia se define como una enfermedad poco común de la infancia que afecta el cerebro y otras partes del cuerpo. Es una inmunodeficiencia primaria que afecta a una variedad de órganos en el cuerpo. Los pacientes con Ataxia-Telangiectasia tienen una marcha inestable (Ataxia), vasos sanguíneos dilatados (Telangiectasia), y una inmunodeficiencia variable que involucra tanto a los linfocitos B como a los linfocitos T(5,6,11,23).
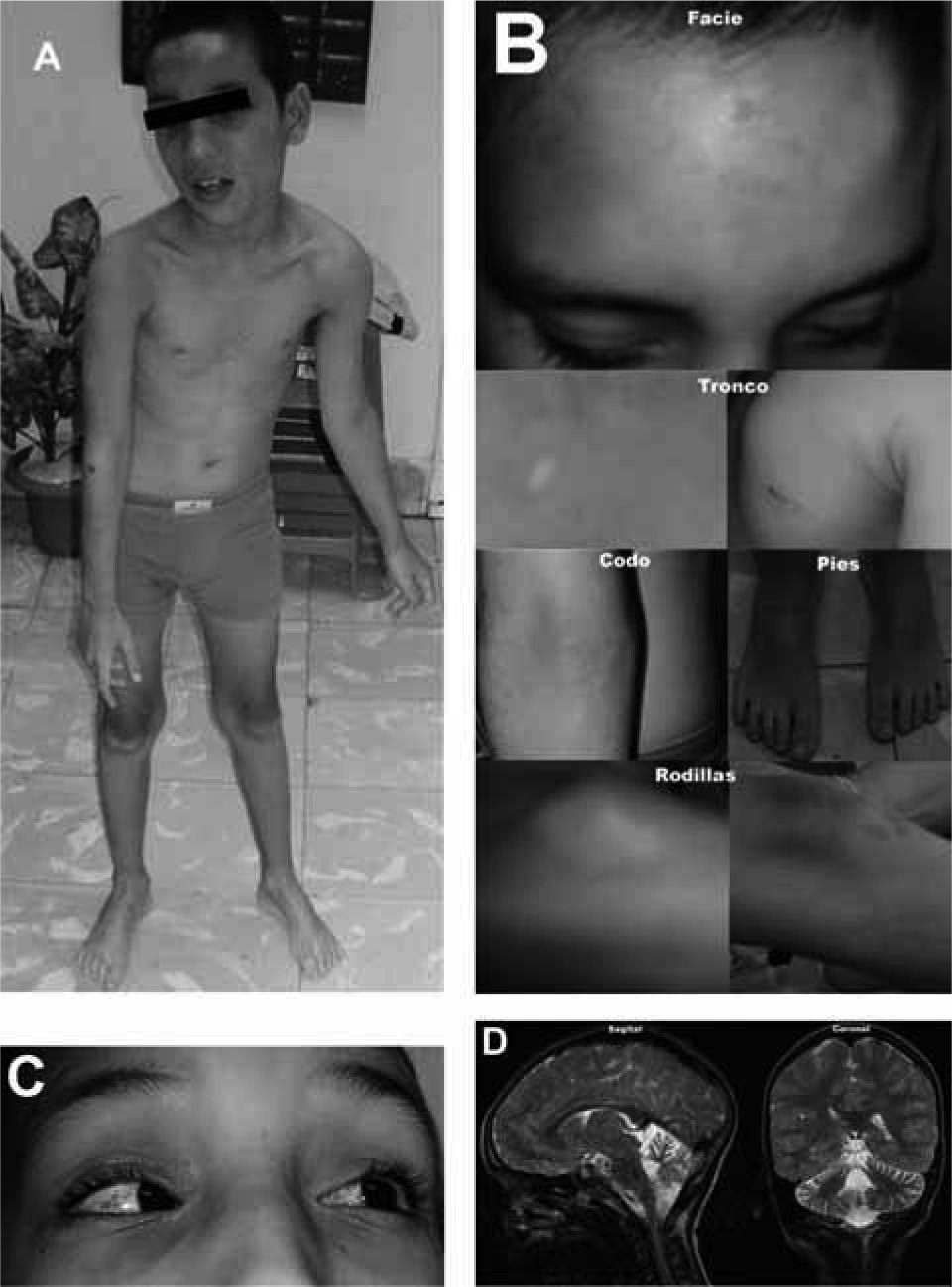
El primer síntoma que se presenta es generalmente ataxia. Los niños con A-T pueden ladearse cuando se paran o se sientan, y balancearse o tambalearse cuando caminan. Esto usualmente resulta de anormalidades neurológicas que afectan la parte del cerebro (cerebelo) que controla el equilibrio. La A-T se vuelve aparente por primera vez cuando el niño empieza a caminar, generalmente entre los 12 y los 18 meses de vida. En esta etapa temprana, se piensa que los niños tienen parálisis cerebral o un trastorno neurológico indefinido. El diagnóstico específico de A-T puede ser difícil de realizar cuando los síntomas aparecen por primera vez. Los síntomas neurológicos posteriores incluyen anormalidades en los movimientos de los ojos, incluyendo contracciones rápidas alternantes de los ojos (nistagmo) y dificultad al iniciar movimientos voluntarios de los ojos (apraxia oculomotora). Presentan también dificultad al usar los músculos necesarios para el habla (disartria) y para deglutir(1,2,6,8). Nuestro paciente, masculino, sin antecedentes familiares aparentes de inmunodeficiencia, se presentó a los 10 años de edad. Comenzó a caminar a los 20 meses, con carencia de equilibrio y movimientos involuntarios. Al examen físico realizado a los 2 años de edad, se constató marcha inestable, ataxia de tronco y de la marcha, con inclinación de la cabeza, hipotonía muscular generalizada y reflejos osteotendinosos exaltados (Fig. 1A). Facies relajada, sonriente, buen humor, con apariencia de máscara, postura de los dedos distónica, sacudidas mioclónicas del tronco y extremidades, disartria, nistagmos, apraxia oculomotora, no retardo mental, si lento en el pensamiento hablado. En piel y mucosas hiperpigmentación e hipopigmentación con atrofia cutánea, manchas color café con leche e hipocrómicas (Fig. 1B).
 Ataxia troncal y de la marcha con inclinación de la cabeza. B) Manchas color café con leche e hipocrómicas. C) Telangiectasias en conjuntiva bulbar. D) Imagen de RMN en la que se aprecian atrofia cerebelosa y dilatación del IV ventrículo.")
La única ataxia asociada a elevación de α-fetoproteína es la Ataxia Telangiectasia, hecho conocido hace más de treinta años(20). En el paciente estudiado los niveles de α-fetoproteína al año de edad, se encontraban elevados: 40.7 UI.
Los vasos sanguíneos dilatados (telangiectasia) se vuelven aparentes después del comienzo de la ataxia, generalmente entre los 3 y los 8 años de edad. La telangiectasia ocurre usualmente en la porción blanca del ojo (conjuntiva bulbar), pero puede encontrarse también en las orejas, cuello y extremidades. En nuestro paciente, a los 3 años de edad, las telangiectasias (Fig. 1C) se hicieron aparentes en conjuntiva bulbar, pabellón de la oreja y tobillos(36).
En la tomografía axial computarizada de cráneo realizada a los 2 años de edad se apreció un aracnoidocele intracelar. Estudio oftalmológico normal, y en el ultrasonido de timo ecogenicidad normal; LD midió 23x10x12 mm y LI midió 26x14x12 mm. El ultrasonido de tiroides de ecogenicidad y dimensiones conservadas. Diseño normal, no se definen adenopatías. De acuerdo a Tavani y colaboradores, la RMN y las tomografías axiales computarizadas realizadas esporádicamente mostraban con frecuencia atrofia cerebelosa no específica y dilatación del IV ventrículo. La atrofia cerebelosa progresa con la edad, comenzando en la infancia temprana(37). La RMN (Fig. 1D) y la tomografía axial computarizada de nuestro paciente mostraron atrofia cerebelosa y dilatación del IV ventrículo.
Otra característica clínica de la A-T es una mayor susceptibilidad a infecciones. Este síntoma es una característica principal en algunos individuos. Las infecciones involucran más comúnmente a los pulmones y senos paranasales, y son usualmente provocadas por bacterias o virus. Las infecciones son, por lo menos en parte, debido a la inmunodeficiencia variable observada en la A-T(9,14,17,38). Otro factor que puede contribuir a las infecciones en pulmón es la disfunción de deglución que resulta en aspiración de alimentos sólidos y líquidos cuando bajan por el tracto a los pulmones (tráquea) en lugar del tracto al estómago (el esófago)(38-40). Nuestro paciente ha presentado episodios frecuentes de infecciones respiratorias altas: rinitis purulenta, sinusitis maxilar; bronconeumonías a repetición y giardiasis frecuentes.
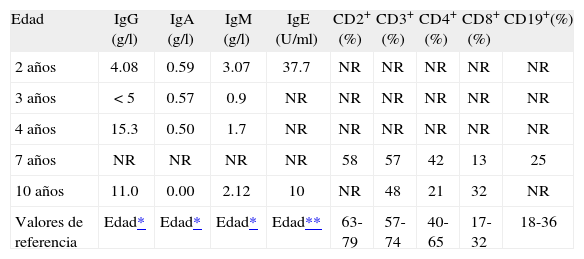
Los pacientes con A-T pueden presentar defectos tanto en el sistema de linfocitos T como en el sistema de linfocitos B. Pueden tener menor cantidad de linfocitos T en sangre. Estas anormalidades en linfocitos T por lo general se encuentran con una glándula, el timo, pequeña o inmadura. La menor cantidad de linfocitos T no siempre aumenta la susceptibilidad del paciente a infecciones(39,41-43). En HH nuestro paciente, el estudio de subpoblaciones linfocitarias efectuado a los 7 años de edad mostró que la proporción de linfocitos T CD2+CD8+ estaban disminuida, con células B periféricas presentes (Tabla I), a los 10 años CD3+CD4+ disminuídos e inversión del índice CD4+/CD8+. La prueba cutánea retardada (a los 8 años de edad) para los antígenos probados (candidina, tricofitina, toxoide tetánico, bacterias respiratorias, estafilococo, estreptococo) midió menos de 5mm para todos los antígenos, por lo que el resultado fue bajo (Tabla II)(9,31,43,44).
Niveles de inmunoglobulinas y subpoblaciones linfocitarias
| Edad | IgG (g/l) | IgA (g/l) | IgM (g/l) | IgE (U/ml) | CD2+ (%) | CD3+ (%) | CD4+ (%) | CD8+ (%) | CD19+(%) |
| 2 años | 4.08 | 0.59 | 3.07 | 37.7 | NR | NR | NR | NR | NR |
| 3 años | < 5 | 0.57 | 0.9 | NR | NR | NR | NR | NR | NR |
| 4 años | 15.3 | 0.50 | 1.7 | NR | NR | NR | NR | NR | NR |
| 7 años | NR | NR | NR | NR | 58 | 57 | 42 | 13 | 25 |
| 10 años | 11.0 | 0.00 | 2.12 | 10 | NR | 48 | 21 | 32 | NR |
| Valores de referencia | Edad* | Edad* | Edad* | Edad** | 63-79 | 57-74 | 40-65 | 17-32 | 18-36 |
NR: No realizado.
Las respuestas desordenadas de anticuerpos pueden estar asociadas con niveles anormales de inmunoglobulinas: IgA ausente en 70% de los pacientes(45), IgE ausente en 80% de los pacientes(46). Las deficiencias de subclases de IgG pueden también ser encontradas en individuos con A-T(17-19,21,47). Nuestro caso presentó, a los 2 años de edad, una disminución de las concentraciones séricas de IgG e IgA, con IgM aumentada, y niveles levemente disminuidos de IgE; a los 3 años de edad una disminución de las concentraciones séricas de IgG e IgA con IgM normal; a los 4 años de edad una disminución de las concentraciones séricas de IgA con IgG e IgM normales, y a los 10 años de edad IgA ausente con IgG e IgM normales y niveles bajos de IgE (Tabla I). La secuencia en la deficiencia de IgA y la inversión del índice CD4+CD8+ es comparable a lo documentado en la literatura(48-50). No existe un patrón constante de defectos inmunológicos en la A-T. Los más notables son el déficit celular (T), la ausencia o disminución de IgA y de subclases de IgG(43,51). En el paciente predominó el defecto de IgA e IgE; sin embargo la IgG, la IgM y las células CD8+ mostraron resultados variables.
Los linfocitos B y los componentes del complemento C3 y C4 fueron normales, como ocurre en los cuatro casos publicados por WC Neves Forte y colaboradores(48).
El ultrasonido de abdomen mostró bazo pequeño, el de cuello tiroides de tamaño normal, no se apreciaron adenopatías, solo un ganglio pequeño de 10mm en diámetro mayor en la región lateral derecha del cuello, el de mediastino no visualizó timo(5,6,17,40,43,52).
Por último, los pacientes con A-T tienen un mayor riesgo a manifestar ciertas clases de cáncer, particularmente del sistema inmune, tal como linfoma y leucemia(2,16,53,54). En este paciente no se han presentado manifestaciones clínicas de cáncer.
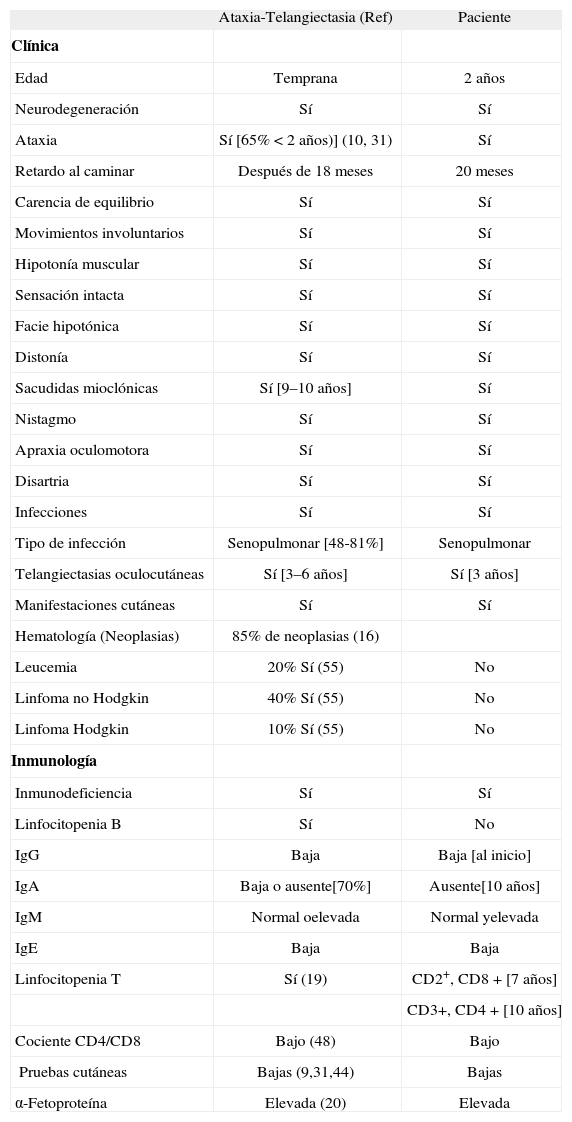
En la Tabla III se comparan los hallazgos clínicos, hematológicos e inmunológicos con los datos de las revisiones realizadas por Boder, Biemond, Centerwall, Chun, Gatti, Gutmann y otros autores(3,9,10,13,16,18-20,31,44,48,55).
Comparación de las alteraciones de la Ataxia Telangiectasia y el paciente al diagnóstico
| Ataxia-Telangiectasia (Ref) | Paciente | |
| Clínica | ||
| Edad | Temprana | 2 años |
| Neurodegeneración | Sí | Sí |
| Ataxia | Sí [65%<2 años)] (10, 31) | Sí |
| Retardo al caminar | Después de 18 meses | 20 meses |
| Carencia de equilibrio | Sí | Sí |
| Movimientos involuntarios | Sí | Sí |
| Hipotonía muscular | Sí | Sí |
| Sensación intacta | Sí | Sí |
| Facie hipotónica | Sí | Sí |
| Distonía | Sí | Sí |
| Sacudidas mioclónicas | Sí [9–10 años] | Sí |
| Nistagmo | Sí | Sí |
| Apraxia oculomotora | Sí | Sí |
| Disartria | Sí | Sí |
| Infecciones | Sí | Sí |
| Tipo de infección | Senopulmonar [48-81%] | Senopulmonar |
| Telangiectasias oculocutáneas | Sí [3–6 años] | Sí [3 años] |
| Manifestaciones cutáneas | Sí | Sí |
| Hematología (Neoplasias) | 85% de neoplasias (16) | |
| Leucemia | 20% Sí (55) | No |
| Linfoma no Hodgkin | 40% Sí (55) | No |
| Linfoma Hodgkin | 10% Sí (55) | No |
| Inmunología | ||
| Inmunodeficiencia | Sí | Sí |
| Linfocitopenia B | Sí | No |
| IgG | Baja | Baja [al inicio] |
| IgA | Baja o ausente[70%] | Ausente[10 años] |
| IgM | Normal oelevada | Normal yelevada |
| IgE | Baja | Baja |
| Linfocitopenia T | Sí (19) | CD2+, CD8+ [7 años] |
| CD3+, CD4+ [10 años] | ||
| Cociente CD4/CD8 | Bajo (48) | Bajo |
| Pruebas cutáneas | Bajas (9,31,44) | Bajas |
| α-Fetoproteína | Elevada (20) | Elevada |
Después de realizado el diagnóstico de inmunodeficiencia se inició tratamiento con Gammaglobulina, 1 bulbo (2ml al 16%) intramuscular cada 21 días durante 6 meses, dieta balanceada, vitaminoterapia y orientación a los padres sobre los cuidados del paciente, con evolución satisfactoria a pesar del pronóstico reservado de este síndrome que se caracteriza por neurodegeneración progresiva.
DECLARACIÓN DE CONFLICTO DE INTERÉSLos autores declaran no tener conflictos de interés.
Los autores agradecen a la Dra. Teresa Español por sus sabios consejos, al paciente R.A. por su disponibilidad y cooperación, y a Alejandro del Valle por las fotos.