


Las inmunodeficiencias humanas del TCR son enfermedades autosómicas recesivas con baja prevalencia, caracterizadas por un defecto de expresión del TCR asociado a una linfopenia T selectiva (más leve en el caso de CD3γ, TCRα o CD247, o grave en el caso de CD3δ o CD3¿). La ausencia congénita de componentes del TCR tiene un impacto diferencial en el desarrollo y función de los linfocitos T, que depende de la cadena del TCR afectada y de la especie, siendo en algunos casos diferente en los pacientes humanos en comparación con los modelos en ratones.
El estudio del inmunofenotipo mediante citometría de flujo, junto con los estudios moleculares, proporciona información esencial para el diagnóstico y el tratamiento, que continúa siendo a día de hoy el trasplante de progenitores hematopoyéticos en los casos asociados a inmunodeficiencia grave.
T-cell receptor (TCR) immunodeficiencies of humans are low-prevalence autosomal recessive diseases characterized by impaired surface TCR expression and selective T lymphopenia (milder in CD3γ, TCRα or CD247 deficiency, and severe in individuals lacking CD3δ or CD3¿). The congenital absence of TCR components has a differential impact on T-cell development and function depending on the affected TCR chain and on the species, with human patients being, in some cases, rather different from mouse counterparts.
The study of the immunophenotype by flow cytometry, along with molecular analyses, provides essential information for diagnosis and treatment, which is still to date the transplant of hematopoietic progenitors in severe immunodeficiency associated cases.
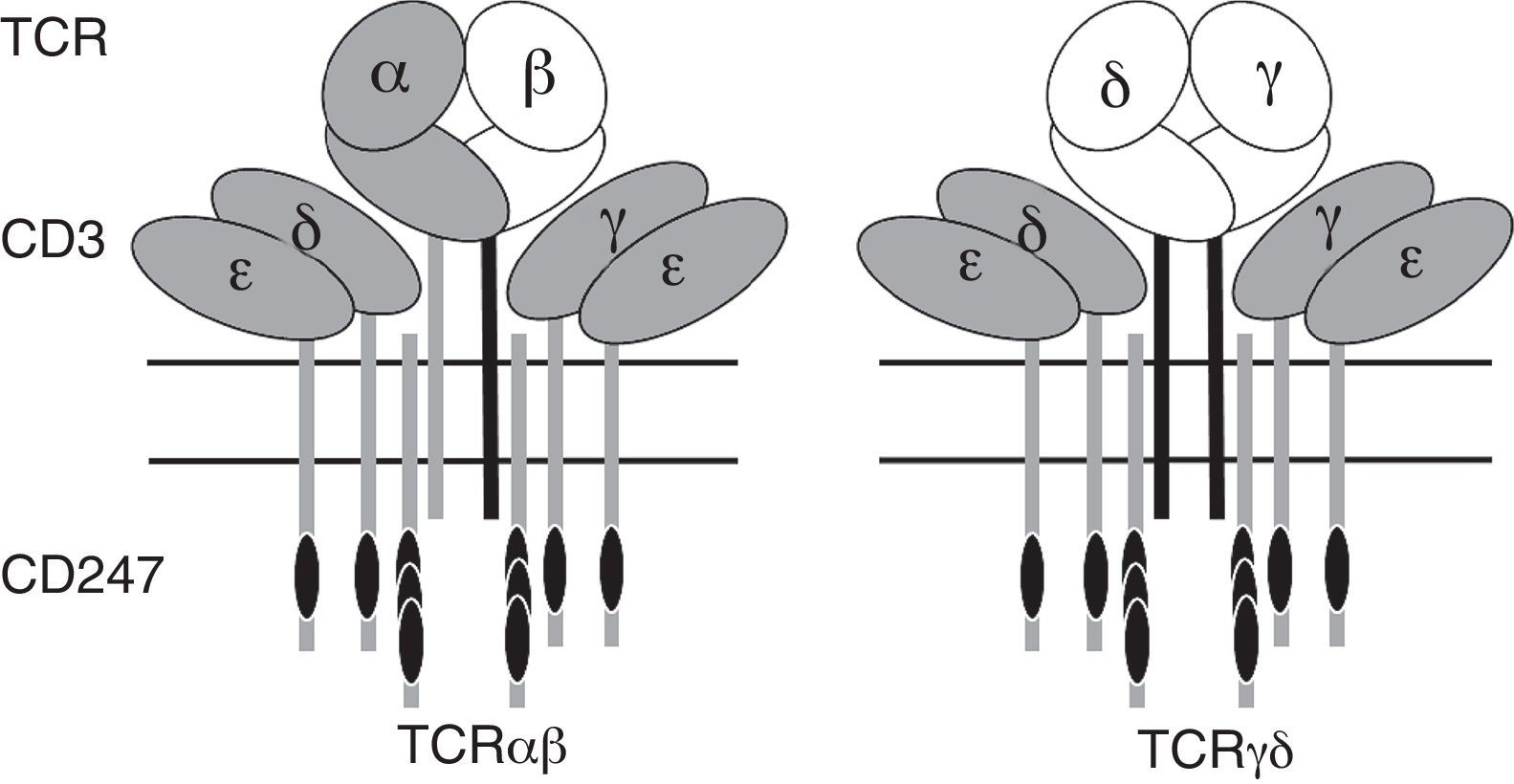
El receptor de antígeno de los linfocitos T (TCR) es un complejo tetradimérico constituido por un heterodímero clonotípico (TCRαβ o γδ) que confiere la especificidad de reconocimiento antigénico, y tres dímeros invariantes: los heterodímeros CD3δ¿ y CD3γ¿ y el homodímero TCRζ o CD247 (fig. 1). Las cadenas invariantes participan en el ensamblaje y expresión en superficie del TCR, y en la transmisión de señales intracelulares para la activación, proliferación y la función efectora o la anergia/apoptosis tras el reconocimiento de antígeno1. Las cadenas invariantes carecen de actividad enzimática intrínseca para la transducción de señales, pero contienen motivos ITAM que, tras su fosforilación, reclutan y unen proteínas cinasas y moléculas adaptadoras como ZAP-70, Lck, LAT, SLP-76, SIT o Nck, las cuales se activan y propagan la señal procedente del TCR2.
 y los homodímeros invariantes CD247 sufren una serie de cambios conformacionales y reclutan enzimas intracelulares (como Fyn, Lck y Zap-70) para iniciar la transducción de la señal. En gris las cadenas para las que se han descrito deficiencias congénitas.")
Isoformas del TCR y sus deficiencias.
Los heterodímeros variables TCR unen antígenos, mientras que los heterodímeros invariantes CD3 (γ¿ y δ¿) y los homodímeros invariantes CD247 sufren una serie de cambios conformacionales y reclutan enzimas intracelulares (como Fyn, Lck y Zap-70) para iniciar la transducción de la señal. En gris las cadenas para las que se han descrito deficiencias congénitas.
Los linfocitos Tαβ reconocen péptidos procesados restringidos por el complejo principal de histocompatibilidad (MHC) o lípidos asociados a CD1 (MHC no-clásico)3, mientras que los linfocitos Tγδ reconocen antígenos restringidos por MHC clásicos, otros por no clásicos y otros se unen directamente al antígeno4. Incluso se piensa que estos linfocitos pueden ser activados para la producción de citocinas, sin necesidad de contacto antígeno-TCR5. Debido al papel fundamental del TCR en la biología del linfocito T, y por extensión en el sistema inmunitario, la descripción en 1986 de la primera deficiencia humana de CD3 (dos casos de la misma familia, uno de ellos sano, [tabla 1] fue una sorpresa6. Cuatro años más tarde se descubrió un tercer caso de deficiencia de CD3 descrita en otro niño7. Poco después se publicó que los primeros casos eran el resultado de una deficiencia selectiva de la cadena CD3γ8, convirtiéndose en la primera inmunodeficiencia congénita del linfocito T de base genética conocida, mientras que el tercer caso era consecuencia de una deficiencia parcial de CD3¿9. Posteriormente, se han descrito nuevas inmunodeficiencias para todas las cadenas invariantes del TCR, e incluso para la cadena variable TCRα (tabla 1), que se pueden clasificar como deficiencias completas (absolutas) o parciales (también conocidas como leaky) en función de los niveles de expresión de la proteína afectada (ausente o reducida, respectivamente).
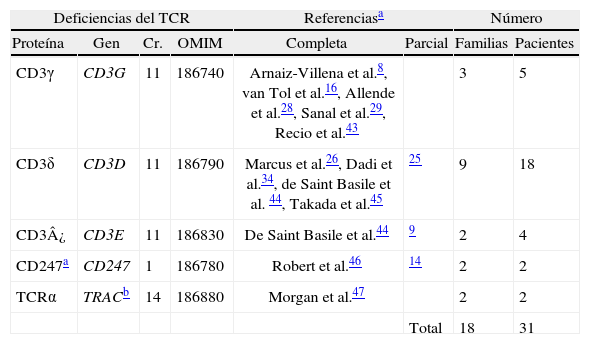
Deficiencias del TCR
| Deficiencias del TCR | Referenciasa | Número | |||||
| Proteína | Gen | Cr. | OMIM | Completa | Parcial | Familias | Pacientes |
| CD3γ | CD3G | 11 | 186740 | Arnaiz-Villena et al.8, van Tol et al.16, Allende et al.28, Sanal et al.29, Recio et al.43 | 3 | 5 | |
| CD3δ | CD3D | 11 | 186790 | Marcus et al.26, Dadi et al.34, de Saint Basile et al. 44, Takada et al.45 | 25 | 9 | 18 |
| CD3¿ | CD3E | 11 | 186830 | De Saint Basile et al.44 | 9 | 2 | 4 |
| CD247a | CD247 | 1 | 186780 | Robert et al.46 | 14 | 2 | 2 |
| TCRα | TRACb | 14 | 186880 | Morgan et al.47 | 2 | 2 | |
| Total | 18 | 31 | |||||
Las inmunodeficiencias del TCR son enfermedades autosómicas recesivas de muy baja prevalencia, que se caracterizan por un defecto de expresión del TCR, frecuentemente asociadas a una linfopenia selectiva de los linfocitos T y a inmunodeficiencia combinada grave (SCID). Son causadas por un amplio rango de mutaciones, cuyos efectos pueden ser completos o parciales, en los genes que codifican las cadenas que componen el TCR. Existen bases de datos de estas mutaciones, con excepción hasta la fecha de aquellas que afectan a las cadenas TCRβ, TCRγ y TCRδ (http://bioinf.uta.fi/base_root/index.php), así como Webs de apoyo diagnóstico (http://bioinf.uta.fi/IDdiagnostics) para la mayoría de estos defectos.
Manifestaciones clínicasEn los últimos años se ha producido un aumento en las inmunodeficiencias del TCR caracterizadas: hasta 30 pacientes descritos en 18 familias en todo el mundo, la mayoría relativas a CD3δ (tabla 1). Las manifestaciones clínicas de estas inmunodeficiencias incluyen una aparición temprana (primer año de vida) y SCID: infecciones respiratorias recurrentes, diarrea crónica y retraso en el desarrollo. En casos descritos para la inmunodeficiencia de CD3γ o CD3¿ parcial algunos individuos presentan signos leves de inmunodeficiencia, aunque no requieren trasplante de médula ósea, ya que los pacientes han alcanzado la tercera década de vida sin necesidad de este. Además, en las inmunodeficiencias de CD3δ, TCRζ y TCRα se han observado características del síndrome de Omenn (hipereosinofilia, hiper-IgE y dermatitis atópica). Se observan infecciones piogénicas, aunque no de carácter crónico. No se observan rasgos dismórficos o anomalías óseas. Por otra parte, la detección de enteropatías en las inmunodeficiencias de CD3δ y CD3γ sugiere una desregulación inmunitaria10.
Las biopsias de tejido linfoide revelan generalmente una reducción de linfocitos, y en las inmunodeficiencias de CD3δ y CD3γ, en particular, es posible la detección del timo. No obstante, a menos que se lleve a cabo un trasplante de médula ósea, la mayoría de los pacientes fallece a temprana edad por infecciones.
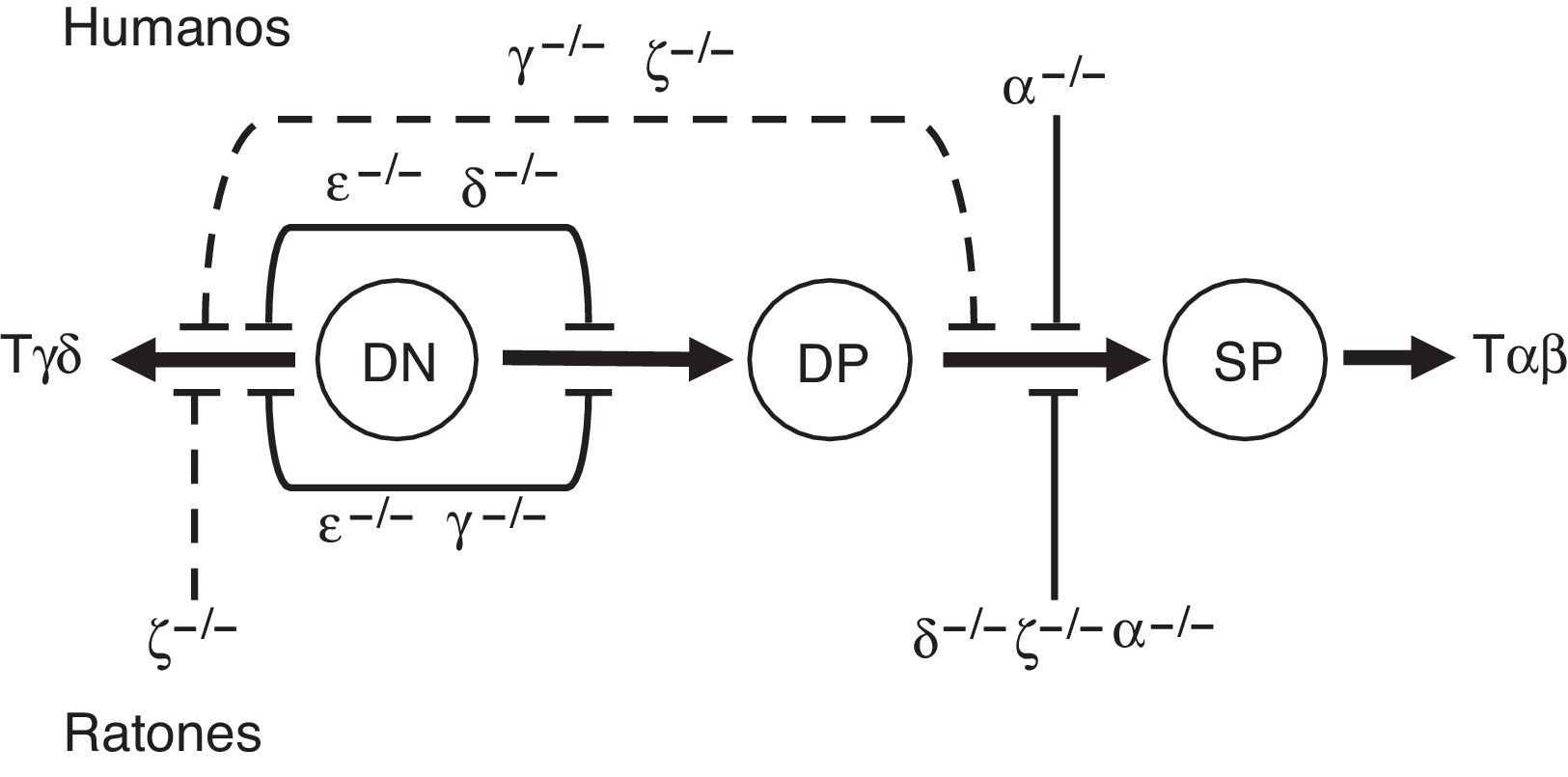
Bases molecularesLa deficiencia específica de alguna de las cadenas que constituyen el TCR tiene un impacto diferencial en la expresión y función de sus distintas isoformas (pre-TCR, TCRαβ y TCRγδ) durante el desarrollo (fig. 2). Estas diferencias son debidas al papel específico de cada una de las cadenas en el ensamblaje o la señalización intracelular iniciada por el receptor.
 o total (—) de la diferenciación temprana de las células T causada por defectos completos de las cadenas del TCR en humanos o ratones. El desarrollo de las células T se simplifica en 2 pasos: 1) DN (CD4-CD8−) a DP (CD4+CD8+) mediado por el pre-TCR, o transición a célula T γδ, respectivamente; y 2) selección positiva/negativa mediada por el TCRαβ y generación de células T αβ SP (CD4+ y CD8+). CD247 y TCRα se han representado como ζ y α, respectivamente.")
Desarrollo de linfocitos T en el timo en las deficiencias completas del TCR.
Bloqueo parcial (- - -) o total (—) de la diferenciación temprana de las células T causada por defectos completos de las cadenas del TCR en humanos o ratones. El desarrollo de las células T se simplifica en 2 pasos: 1) DN (CD4-CD8−) a DP (CD4+CD8+) mediado por el pre-TCR, o transición a célula T γδ, respectivamente; y 2) selección positiva/negativa mediada por el TCRαβ y generación de células T αβ SP (CD4+ y CD8+). CD247 y TCRα se han representado como ζ y α, respectivamente.
El modelo de ensamblaje actualmente aceptado está basado en una jerarquía de asociación de los dímeros clonotípicos (TCRαβ y TCRγδ) con los dímeros invariantes (CD3δ¿, CD3γ¿, y TCRζζ)11: el dímero CD3δ¿ se une a la cadena TCRα, y solo entonces se produce la asociación del dímero CD3¿γ a la cadena TCRβ, dando lugar al hexámero TCRαβ/CD3¿δ¿γ. Por último, la incorporación del homodímero TCRζζ resulta en un complejo completo de estequiometría TCRαβ/CD3¿δ¿γ/TCRζ2. Así, dado el requerimiento diferencial de los componentes del TCR para su ensamblaje (lo que condiciona su expresión y función), el fenotipo de la inmunodeficiencia puede variar entre un bloqueo temprano y absoluto en el desarrollo, como el observado en las deficiencias de CD3δ o CD3¿, o un bloqueo parcial, como en el caso de las cadenas CD3γ o TCRζ (fig. 2).
Diagnóstico diferencialEl estudio del inmunofenotipo mediante citometría de flujo, junto con los estudios moleculares, proporciona información esencial para el diagnóstico y la clasificación de las inmunodeficiencias del TCR.
InmunofenotipoUno de los hallazgos más consistentes en las inmunodeficiencias del TCR es la linfopenia T selectiva y persistente, la cual permite clasificar a los pacientes deficientes en dos grupos. El primer grupo se caracteriza por una linfopenia T grave (<2% de linfocitos T periféricos, inmunofenotipo T−B+NK+), que es lo observado en las deficiencias absolutas de CD3¿ o CD3δ. La linfopenia total (<3.000 linfocitos/μl en niños) es frecuente en este grupo, salvo en algunos casos en los que se compensa por la expansión de los linfocitos B y NK. En el segundo grupo la linfopenia T es leve (>20%, inmunofenotipo T+/−B+NK+), como se observa en las deficiencias completas de CD3γ, TCRζ y TCRα, así como también en la mayoría de las deficiencias parciales (tabla 2).
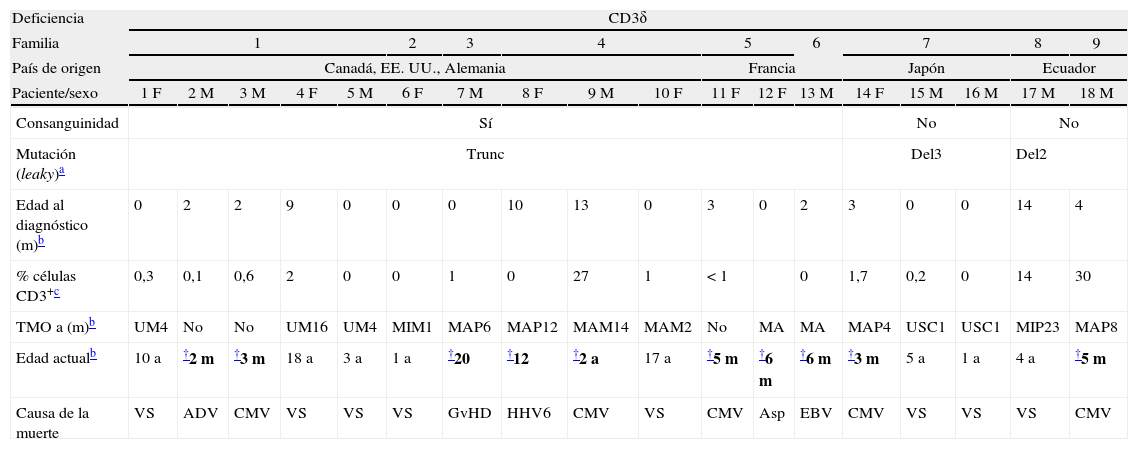
Deficiencias del TCR. Datos clínicos e inmunológicos
| Deficiencia | CD3δ | |||||||||||||||||
| Familia | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |||||||||
| País de origen | Canadá, EE. UU., Alemania | Francia | Japón | Ecuador | ||||||||||||||
| Paciente/sexo | 1F | 2M | 3M | 4F | 5M | 6F | 7M | 8F | 9M | 10F | 11F | 12F | 13M | 14F | 15M | 16M | 17M | 18M |
| Consanguinidad | Sí | No | No | |||||||||||||||
| Mutación (leaky)a | Trunc | Del3 | Del2 | |||||||||||||||
| Edad al diagnóstico (m)b | 0 | 2 | 2 | 9 | 0 | 0 | 0 | 10 | 13 | 0 | 3 | 0 | 2 | 3 | 0 | 0 | 14 | 4 |
| % células CD3+c | 0,3 | 0,1 | 0,6 | 2 | 0 | 0 | 1 | 0 | 27 | 1 | <1 | 0 | 1,7 | 0,2 | 0 | 14 | 30 | |
| TMO a (m)b | UM4 | No | No | UM16 | UM4 | MIM1 | MAP6 | MAP12 | MAM14 | MAM2 | No | MA | MA | MAP4 | USC1 | USC1 | MIP23 | MAP8 |
| Edad actualb | 10 a | †2 m | †3 m | 18 a | 3 a | 1 a | †20 | †12 | †2 a | 17 a | †5 m | †6 m | †6 m | †3 m | 5 a | 1 a | 4 a | †5 m |
| Causa de la muerte | VS | ADV | CMV | VS | VS | VS | GvHD | HHV6 | CMV | VS | CMV | Asp | EBV | CMV | VS | VS | VS | CMV |
| Deficiencia | CD3γ | CD3¿ | TCRζ | TCRα | |||||||||
| Familia | 1 | 2 | 3 | 1 | 2 | 1 | 2 | 1 | 2 | ||||
| País de origen | Turquía | España | Francia | Caribe Hawai | Pakistán | ||||||||
| Paciente/sexo | 1M | 2M | 3M | 4M | 5M | 1M | 2F | 3M | 4F | 1M | 2F | 1F | 2M |
| Consanguinidad | Sí | No | No | Sí | ND | No | Sí | ||||||
| Mutación (leaky)a | Trunc | Trunc, Het | Del7, Trunc, Het | Trunc | Trunc | Ins | Del3 | ||||||
| Edad al diagnóstico (m)b | 3 | 7 | 48 | 12 | 48 | 48 | 5 | 1 | 0 | 4 | 10 | 15 | 6 |
| % células CD3+c,d | 37 | 27 | 30 | 10 | 18 | 63 | ND | ND | <1 | 21 | 64 | 21 | 50 |
| TMO a (m) b | No | MI | No | No | No | No | No | No | MA | MA | MA | MIM6a | MIM7a |
| Edad actualb | †9 m | †20 m | 20 a | †32 m | 30 a | 22 a | †5 m | †3 m | †2 m | 10 a | 12 a | 9 a | 10 a |
| Causa de la muerte | Sepsis | Neum | VS | CMV | VS | VS | Neum | CMV | ADV | VS | VS | VS | VS |
ADV: Aleutian disease virus; Asp: Aspergillus; CMV: citomegalovirus; Del7: deleción del exón 7; EBV: virus de Epstein Barr; GvHD: enfermedad injerto contra huésped; Het: heterocigoto compuesto; HHV: herpesvirus humano; Ins: inserción; ND: no descrito; Neum: neumonía; M: médula ósea; MA: HLA matched related donor; MI: HLA missmatched related; P: sangre periférica; SC: sangre de cordón umbilical; Trunc: proteína truncada; U: matched unrelated donor;VS: vivo y sano.
El defecto de expresión del TCR, cuando existen linfocitos T en periferia, es otro de los hallazgos característicos en este tipo de inmunodeficiencias. La pérdida de expresión se detecta y cuantifica con anticuerpos monoclonales anti-CD3 mediante citometría de flujo. La expresión es de 2 a 10 veces menor en los individuos inmunodeficientes que en los controles normales. La pérdida de expresión igual o superior a 10 veces, considerada como muy grave, se observa en las inmunodeficiencias completas de TCRζ, TCRα y CD3¿ parcial. Sin embargo, en las inmunodeficiencias de CD3γ y CD3δ parcial la expresión es solo 5 veces menor que en los controles.
Por último, la capacidad de exportación de linfocitos T maduros desde el timo puede ser evaluada a partir del repertorio TCRVβ mediante citometría de flujo o PCR cuantitativa12, la cuantificación y monitorización del número de círculos de escisión del receptor de linfocitos T (T-cell receptor excision circles [TREC])13, o la identificación de emigrantes tímicos con el inmunofenotipo CD45RA+CD31+. Los pacientes inmunodeficientes se caracterizan por un repertorio TCRVβ restringido y un reducido número de TREC, lo que sugiere un aporte deficitario desde el timo.
Diagnóstico molecularEl análisis molecular se realiza por secuenciación de los genes codificantes de las cadenas del complejo CD3, CD247 y TRAC. Los genes pueden contener mutaciones que, dependiendo de su localización, generan efectos cuantitativos y cualitativos diferentes, que pueden manifestarse como una carencia total o parcial de actividad del TCR (dependiendo del grado de expresión de la proteína afectada). Las deleciones se deben a mutaciones en sitios de ayuste (splicing), y fueron identificadas en el ADN genómico mediante secuenciación entre los exones de interés. Como consecuencia se trunca la proteína o se pierde el exón. Curiosamente, en ciertas inmunodeficiencias, como la deficiencia parcial de TCRζ, se ha observado una reversión en el defecto de expresión del TCR debido a mutaciones adicionales en los precursores de las células T14. Por último, la identificación de las mutaciones facilita el diagnóstico directo de la inmunodeficiencia mediante oligonucleótidos específicos, enzimas de restricción o secuenciación directa.
Modo de herencia e identificación de portadoresLas inmunodeficiencias del TCR son enfermedades autosómicas recesivas con baja prevalencia. Los portadores (heterocigotos) son clínicamente sanos y no se distinguen fácilmente de los individuos normales en la pruebas de laboratorio, aunque a menudo presentan niveles intermedios de expresión en la superficie del TCR detectable por citometría de flujo15 o técnicas bioquímicas16. No obstante, la detección de portadores requiere el análisis molecular para determinar las mutaciones en cada caso.
Estudios funcionalesEl análisis funcional de las células T y B es útil para el diagnóstico de esta clase de inmunodeficiencias. La respuesta funcional T (respuesta frente a anti-CD3 y PHA) o B (respuesta de anticuerpos a la infección o vacunación) es generalmente nula o baja. Sin embargo, los niveles de inmunoglobulinas son normales. La función citolítica de las células NK es normal, excepto para la inmunodeficiencia de TCRζ.
Función in vitroLa función del TCR no puede ser analizada, obviamente, en los pacientes con inmunodeficiencia del TCR que presentan un bloqueo total de la diferenciación T. En los casos de linfopenia T parcial sigue siendo difícil la comparación con individuos sanos, debido al bajo número y a la expresión reducida del TCR en los linfocitos T maduros. En cualquier caso, la sola presencia de linfocitos T en los pacientes sugiere un cierto grado de selección tímica. Por otro lado, en algunos casos (CD3γ, CD3¿ parcial) la respuesta de anticuerpos indica que la función T colaboradora es normal.
Nuestros estudios en la inmunodeficiencia humana de CD3γ usando líneas de linfocitos T (dependientes de IL-2) transformadas con el virus Herpesvirus saimiri o HTLV-I, indican que la cadena CD3γ contribuye, aunque no es absolutamente necesaria, a la regulación del tráfico del TCR17. Asimismo, CD3γ es prescindible para diferentes respuestas funcionales como el flujo de calcio, citólisis, inducción o modulación de moléculas de superficie, proliferación, síntesis de TNFα e IFNγ, y la señalización proximal por ciertas rutas (ZAP-70, ERK, p38 y mTOR) en linfocitos T, y para el desarrollo y expansión de los linfocitos iNKT18. En contraste, en los linfocitos T carentes de CD3γ la internalización del TCR inducida por PMA, la síntesis de IL-2 y la fosforilación de TCRζ se encuentran afectadas8,18–21. La ausencia de la cadena CD3γ reduce la expresión del TCR de forma más marcada en linfocitos CD8+ que en CD4+, tanto en humanos como en ratones. En nuestro laboratorio hemos demostrado que estas diferencias se deben, al menos en parte, a la existencia de diferencias en la glucosilación de las cadenas TCRαβ y CD3δ que determinan su conformación en cada linaje22,23. Por último, la deficiencia de CD3γ afecta más la expresión del TCRαβ que la del TCRγδ,24, mientras que curiosamente la deficiencia parcial de la cadena CD3δ causa el efecto contrario25.
Tratamiento y pronósticoTrasplante de progenitores hematopoyéticosEl trasplante de progenitores hematopoyéticos (TPH) constituye el tratamiento más adecuado para las inmunodeficiencias del TCR, dado que el pronóstico es muy desfavorable en el caso de no realizarse, salvo para la deficiencia de CD3γ o la mayoría de las deficiencias parciales (tabla 2). Los progenitores hematopoyéticos pueden ser obtenidos a partir de médula ósea, sangre periférica o cordón umbilical de donantes genotípicamente idénticos (HLA matched related), parcialmente idénticos (HLA missmatched related), o compatibles pero no emparentados (matched unrelated). Los receptores son sometidos a un condicionamiento mielo-ablativo y el éxito del tratamiento depende de la eficacia de dicha ablación.
Todos los pacientes presentan clínica de inmunodeficiencia, aunque en algunos casos sea de manera leve y en otros de manera severa, hasta el punto de necesitar TPH. El origen de variación en la clínica de estos pacientes sigue aún sin ser aclarado.
En la deficiencia de CD3δ se ha demostrado que el trasplante con donantes no emparentados da mejor resultado que los trasplantes parcialmente idénticos26. Las complicaciones infecciosas relacionadas con la inmunosupresión (Herpesvirus) son la causa más común de muerte entre los pacientes trasplantados. No obstante, todos los pacientes trasplantados con éxito han llevado una vida normal hasta los 18 años postrasplante.
En determinados casos algunos pacientes presentan signos leves de inmunodeficiencia, y por tanto no se consideraba necesario el TPH, como sucede en las inmunodeficiencias de CD3γ y CD3¿ parcial, donde los afectados han alcanzado la tercera década de vida. Sin embargo, el tratamiento profiláctico de elección es la inmunoglobulina intravenosa (IVIG) con o sin antibióticos16,27, si los signos clínicos reaparecen28. En el caso de la inmunodeficiencia de CD3γ la respuesta de anticuerpos fue normal in vivo, por lo que se realizó un programa de vacunación preventivo, excluyendo virus atenuados. Asimismo, este paciente en particular presentaba asma bronquial que se trató con ketotifeno y cromoglicato sódico entre los 3,5 y 7 años29. A partir de los 7 años se trató la hiperreactividad no atópica de las vías respiratorias. El paciente español con deficiencia de CD3γ ha sido tratado con antibióticos solo cuando desarrollaba signos clínicos de inmunodeficiencia, y ha alcanzado la tercera década de vida sin dificultades.
Terapia génicaLos protocolos de terapia génica consisten en la sustitución del gen defectuoso en enfermedades de tipo monogénico. No obstante, deben ser probados previamente in vitro30. En la inmunodeficiencia de CD3γ la expresión de la cadena normal en los linfocitos T maduros restaura la expresión del TCR31. Se observó, sin embargo, un desajuste funcional en los linfocitos primarios, reflejado en la síntesis constitutiva de IL-2 y una aparente autorreactividad.
Modelos animalesEl estudio de ratones con mutaciones de los genes que codifican las cadenas invariantes y variables del TCR ha permitido la caracterización del papel de cada cadena1,32, en particular durante el desarrollo intratímico, y su comparación con pacientes humanos (fig. 2). Este análisis sugiere que, en ciertos aspectos, los requerimientos de dichas cadenas son diferentes en las 2 especies. De hecho, la cadena CD3δ no es necesaria para el desarrollo de las células Tγδ en ratones33, pero sí lo es en humanos34, debido a la distinta estequiometría del TCRγδ humano, que incluye CD3δ, en comparación al TCRγδ del ratón que no incorpora esta cadena24,35 (fig. 1). En contraste, la falta de la cadena CD3δ da lugar al bloqueo completo del desarrollo de los linfocitos Tαβ tanto en humanos como en ratones, aunque en distintos estadios del desarrollo intratímico33,34 (fig. 2). Así, en humanos pero no en ratones36, CD3δ es esencial para la expresión funcional del pre-TCR y la consecuente transición DN-DP durante el desarrollo intratímico Tαβ temprano. Curiosamente, la situación se invierte en el caso de la deficiencia de CD3γ, la cual afecta a la expresión funcional del pre-TCR en el ratón37 pero no en los humanos8 (fig. 2). Estas diferencias establecen limitaciones en cuanto al uso de modelos múridos para la obtención de información relevante en el contexto de las deficiencias humanas del TCR. Una manera de atacar este problema ha sido la generación y análisis de ratones «humanizados» portadores de cadenas TCR38 o CD3 transgénicas de origen humano33,39–41.
En el ratón la deficiencia completa de las distintas cadenas CD3 ha demostrado que el desarrollo de células T se ve alterado de manera drástica en todas las situaciones, aunque en diferentes puntos de control y en un grado diferente (fig. 2). Sin embargo, todas las proteínas CD3 excepto CD3δ son necesarias para la selección β mediada por el pre-TCR. Así, contrariamente a lo propuesto en humanos, la jerarquía CD3 en ratones sería CD3γ mayor que CD3δ.
Además, se ha observado que la expresión incompleta de complejos TCR de ratón en células no T (3T3) es posible cuando CD3δ o γ (pero no CD3¿ o cualquier otra cadena) están ausentes42, apoyando la jerarquía ¿ > γ > δ en ratón.
Cabe destacar aquí la ausencia de CD3δ en las células TCR γδ de ratón que presentan una estequiometría γδ/γ¿γ¿ζζ. Por lo tanto, en células humanas, pero no en ratón, si carecen de CD3δ no presentan ni timocitos dobles positivos ni células Tγδ. Por el contrario, los ratones pero no los seres humanos que carecen de CD3γ tienen muy pocas células T αβ y γδ periféricas.
Tomados en conjunto, estos resultados sugieren un papel diferente para CD3γ y CD3δ en humanos y ratones en el pre-TCR y la función del TCR durante el desarrollo de las células T.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Nuestro trabajo en este campo ha sido financiado por Comunidad Autónoma de Madrid (S2011/BMD-2316), Fundación Lair, Instituto de Salud Carlos III (ISCIII–PI080921 and PI060057, RIER RD08-0075-0002 and PI11/02198), Instituto de Investigación Hospital 12 de Octubre, Ministerio de Ciencia e Innovación (SAF 2011-24235) y Fundación Mutua Madrileña. Agradecemos a los doctores Hidetoshi Takada (Department of Pediatrics, Graduate School of Medical Sciences, Kyushu University), Juana Gil (Inmunología, Hospital Gregorio Marañón, Madrid, España), Eduardo López-Granados (Inmunología, Hospital La Paz, Madrid, España), Chaim M. Roifman (The Canadian Centre for Primary Immunodeficiency, Div. of Immunology and Allergy, The Hospital for Sick Children, Toronto, Ontario, Canadá) y Françoise Le Deist (CHU Sainte-Justine, Montréal, Canadá) sus datos sobre los pacientes y su continua colaboración científica.
Ambos autores han contribuido y han participado por igual y en la misma proporción en la elaboración del presente manuscrito.