


Recent studies have suggested that autophagy can act as a protective immune mechanism against Listeria monocytogenes infection. L. monocytogenes is a Gram-positive, facultative intracellular bacterium that causes invasive diseases in humans and animals, particularly in the central nervous system (CNS). Human listeriosis of the CNS can manifest in many ways, including meningitis and brain abscesses. The initial line of defence against bacterial colonisation is provided by microglia, resident phagocytes of the CNS parenchyma. Microglial cells are also well known for clearing dead and dying neural cells after injury, and therefore play a key role in infectious diseases and neurodegeneration.
Little is known about the role of the autophagy pathway in host–pathogen interactions in the brain as most in vitro studies have used macrophages or epithelial cells to study this interaction. In the present work, a quantitative real time-PCR array analysis was performed to assess autophagy-related gene expression in a brain rat ex vivo organotypic nervous system model during L. monocytogenes infection. We found that, in brief, core autophagy gene expression is not modulated by the infection, despite the presence of intense microglial phagocytic activity on the brain tissue surface that can be seen by scanning electron microscopy. We conclude that, in our model, autophagy could play a role in homeostasis in the damaged brain tissue instead of an immune-relevant pathway.
Estudios recientes han evidenciado que la autofagia puede actuar como un mecanismo inmune protector frente a la infección con Listeria monocytogenes. L. monocytogenes es una bacteria grampositiva, intracelular facultativa, que causa enfermedades invasivas en humanos y animales, especialmente en el sistema nervioso central (SNC). La listeriosis humana en el SNC puede manifestarse de diferentes maneras, incluyendo meningitis y abscesos cerebrales. La línea principal de defensa frente a las infecciones bacterianas es proporcionada por la microglía, fagocitos residentes del parénquima del SNC. Las células de microglía son conocidas, también, por eliminar las células dañadas o muertas tras un daño cerebral, y por lo tanto desempeñan un papel clave en las enfermedades infecciosas y neurodegenerativas.
Se sabe poco sobre el papel de la autofagia en las interacciones entre el hospedador y el patógeno, debido a que la mayoría de los estudios in vitro han usado macrófagos o células epiteliales. En el presente trabajo hemos utilizado matrices de PCR en tiempo real para analizar la expresión de genes de autofagia en un modelo organotípico de cerebro de rata infectado con L. monocytogenes. Hemos observado que, en general, la expresión de genes centrales de la autofagia no está modulada por la infección, a pesar de la presencia de una intensa actividad fagocítica de la microglía en la superficie del tejido cerebral, observada mediante microscopia electrónica de barrido. Concluimos que, en nuestro modelo, la autofagia podría desempeñar un papel clave en la homeostasis del tejido dañado en lugar de tener un papel inmune relevante.
Autophagy is a fundamental homeostatic process in which cellular cytoplasmic targets are sequestered within double-membraned autophagosomes and subsequently delivered to lysosomes for degradation.1 In addition to the degradation of damaged organelles, autophagy has received attention as a crucial component of innate defence against a variety of infectious agents, including parasites, viruses and bacteria.2–4 Therefore, the many presently recognised roles of autophagy in innate and adaptive immunity have been steadily increasing in complexity.5,6 Autophagy of virulent pathogens has emerged as a powerful method of eliminating intracellular bacteria while the exact mechanism of bacterial recognition by autophagy remains unknown.7,8 Moreover, the role of autophagy in bacterial elimination by microglia, the resident immune cells in the central nervous system (CNS), is much less clear.
Listeria monocytogenes, a Gram-positive, facultative intracellular bacterium, is responsible for severe foodborne infections in humans. More than 50% of cases correspond to septicaemia, and around 25% to CNS infections.9 This pathogen has the ability to cross the intestinal, placental and blood–brain barriers.10 Listeriosis occurs primarily in immunocompromised individuals, causing septicaemia, brain abscesses, meningitis; and spontaneous abortion in pregnant women. L. monocytogenes has been shown to interact with the host autophagic machinery.11,12 However, most studies on Listeria and autophagy have been carried out using epithelial or macrophage cell lines, not neurons or microglia. Microglial activation is a key factor in the defence of the neural parenchyma against infectious diseases, inflammation, trauma, ischaemia and neurodegeneration. Recently, we used a CNS model to study the interactions between L. monocytogenes and microglia ex vivo.13 In this model, cycles of microglial phagocytosis against L. monocytogenes and necrotic cells were clearly shown, accompanied by a strong expression of immune relevant genes.
As autophagy eliminates intracellular pathogens in a process similar to unwanted or damaged intracellular organelles, and some immune defence components induce or increase autophagy, it is important to study the cooperation between immune and autophagic genes. In the present work, quantitative RT-PCR array analyses were performed to study autophagy-related gene expression in a rat ex vivo organotypic nervous system model.
Materials and methodsBacterial strainL. monocytogenes strain HUMV-4251 was isolated from human cerebrospinal fluid at the Hospital Universitario Marqués de Valdecilla, Santander, Spain. Species-specific identification was confirmed by partial 16S ribosomal DNA gene sequencing.
Organotypic culturesOrganotypic explants were obtained from newborn (7–10-day-old) Sprague Dawley rats. Their brains were carefully and quickly removed and placed in a Petri dish with Dulbecco's Modified Eagle Medium, on a cold plate to reduce metabolic activity and to facilitate smooth slicing. Brain slices, 300-μm thick, were collected using a vibrating microtome (Microm HM 650V; Thermo Scientific). The cultures were kept in six-well-plates at 37°C in a 5%-CO2-enriched atmosphere for at least one week before bacterial infections. The culture medium was a mixture of Basal Medium Eagle 1× (50%, Gibco-Invitrogen), normal horse serum (25%, Gibco-Invitrogen) and Hank's solution (25%, Gibco). Glutamine (200mM), d-glucose (3.7mM, Sigma), and the antibiotics penicillin, streptomycin and amphotericin b (Gibco-Invitrogen) were also added. The medium was filtered using a 0.2-μm pore sterile filter (Millipore). All animal care and experimental procedures were according to the Spanish legislation and the European Communities Council Directive on the “Protection of Animals Used in Experimental and Other Scientific Purposes” (86/609/EEC).
Bacterial infectionsL. monocytogenes was grown on blood agar plates and cultured overnight in 10mL of Brain Heart Infusion (BHI) Broth. The suspension was centrifuged for 4min at 5000rpm and then resuspended in cell culture medium with a final concentration of ∼1010cfumL−1. The number of colony forming units (CFUs) was determined by serial dilution in phosphate buffered saline (PBS), and plating onto BHI plates. The organotypic culture medium was replaced with fresh, antibiotics-free medium at least 1h before infections. Tissue cultures were infected on the surface with 2μl of medium containing 107 bacteria. Controls were inoculated with cell culture medium alone.
Scanning electron microscopy (SEM)Brain organotypic cultures infected with L. monocytogenes recovered at 5 and 12h, and the respective controls were fixed in ice-cold 3% glutaraldehyde for 20min at 4°C. Samples were then dehydrated in a series of graded acetone solutions, dried by the critical point method, coated with gold in a fine coat ion sputter JFC-1100 (JEOL) and observed with an Inspect S microscope (FEI Company) working at 15 or 20kV.
RNA isolation and cDNA synthesisTotal RNA was extracted from four independent infected organotypic cultures and three controls at each time point (5 and 12h) using TRIzol (Invitrogen). The RNA concentration was quantified with a NanoDrop spectrophotometer and RNA quality was checked by electrophoresis on 1.5% agarose gel. For q-PCR arrays, the RNA was cleaned after ethanol precipitation and cDNA was generated from 400ng of the total RNA using the SABiosciences's RT2 First Strand Kit, according to the manufacturer's protocol (Qiagen), including the DNase treatment step.
Gene expression by q-PCR cDNA arrays and statistical analysisGene expression quantification was performed using a rat autophagy array (PARN-084, SABiosciences-Qiagen). This kit profiles the expression of 84 genes involved in autophagy (Table S1). The array includes genes that encode components of the molecular machinery and key regulators modulating autophagy in response to both extracellular and intracellular signals, genes involved in autophagic vacuole formation, genes responsible for protein targeting to membrane and protein transport, protein ubiquitination, genes linking autophagosomes with lysosomes, co-regulators of autophagy and apoptosis, and autophagy genes induced by intracellular pathogens (Table S2). Moreover, the array includes five reference genes namely ribosomal protein P1 (Rplp1), hypoxanthine phosphoribosyltransferase 1 (Hprt1), ribosomal protein L13A (Rpl13a), lactate dehydrogenase A (Ldha), and beta actin (Actb).
Amplification and data acquisition were carried out by means of the CFX-Manager software (BioRad). The PCR cycling programme was set as follows: stage 1: 95°C for 10min, stage 2: 95°C for 15s followed by 60°C for 1min, repeated for 40 cycles. The threshold cycle (Ct) of each gene was automatically established and recorded by the software.
The delta Ct (ΔCt) method was used for PCR array data analysis. The normalised (ΔCt) for each gene of interest (GOI) was calculated by subtracting the average Ct of the five housekeeping genes from the Ctof each GOI. Next, the double delta Ct (ΔΔCt) for each GOI was calculated by deducting the average ΔCt of GOI in the sham group from the ΔCt of each GOI. The fold-change of each GOI compared with the sham group was calculated as 2ΔΔCt. Ct data were uploaded into the data analysis template on the manufacturer's website (http://www.sabiosciences.com/pcr/arrayanalysis.php) and the p values were calculated based on a Student's test of the replicate (2ΔΔCt) values for each gene in the control and infected groups.
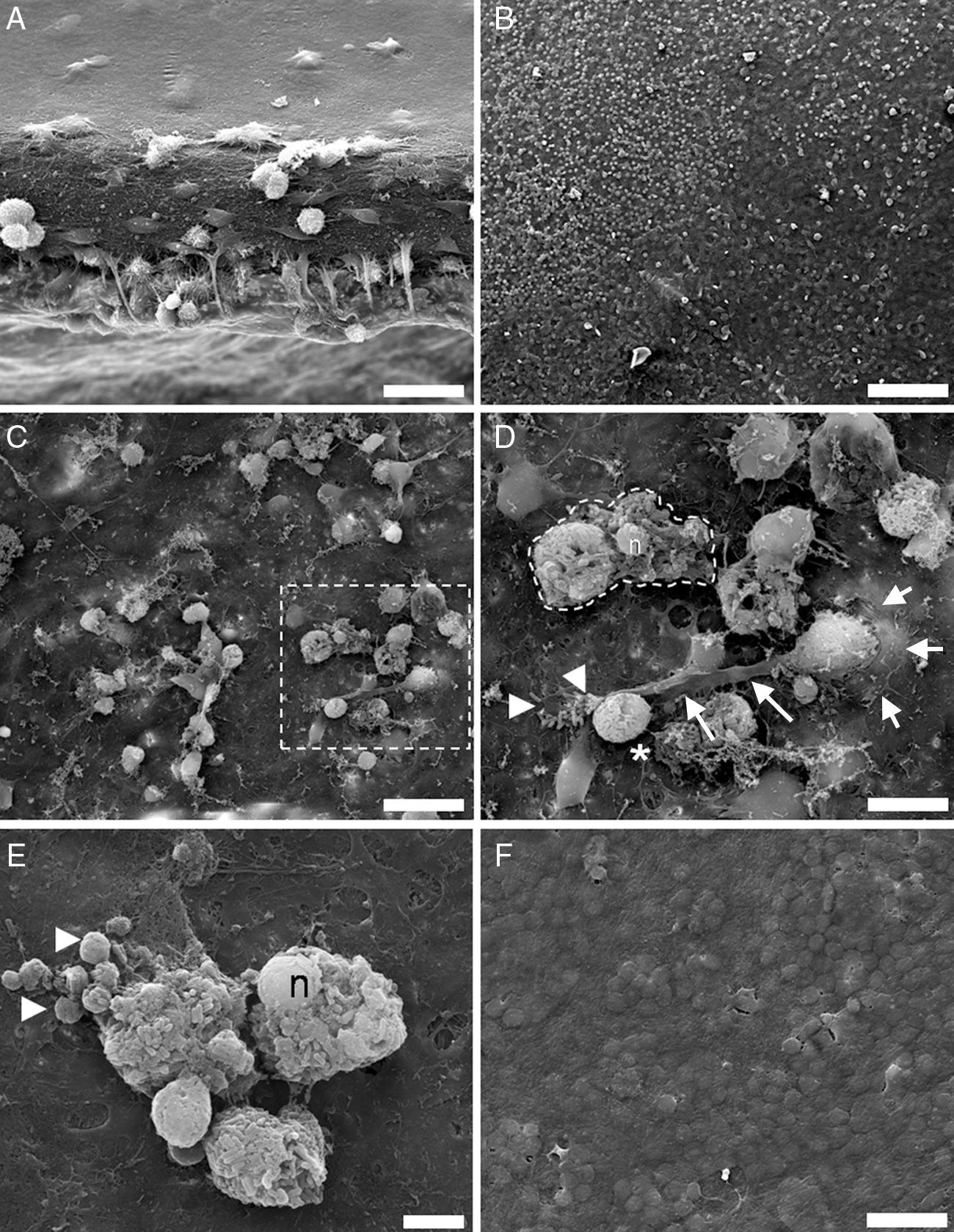
ResultsBefore L. monocytogenes infection, under SEM, the surface of the brain slices appeared to be formed by extremely flat cells (Fig. 1A). During Listeria infection, the smooth surface was disrupted and many microglial cells coming from the inside of the tissue were massively recruited to the surface of the brain slices (Fig. 1B). Five hours after the infection, the explant surface showed microglial activated cells and cells filled with bacteria. New phagocytic cells reappeared on the surface of the organotypic cultures to phagocyte free bacteria and cellular debris (Fig. 1C and D). Cell activity increased on the explant surface 12h after infection, and microglial cells were engulfing infected cells, phagosome-like bodies released from dead microglial cells, and free bacteria. Often, the plasma membrane of infected cells lost its integrity, cytosolic and organellar contents were released into the surrounding environment, and the nuclei were not fragmented (Fig. 1E). Control tissues had a normal appearance with no detectable phenotypic changes (Fig. 1F).
 Shows the thickness of an organotypic culture, with the surface covered cells forming a flat surface. (B) Activation of microglia by L. monocytogenes shows numerous round cells which emerge from the tissue. (C) 5h after infection, bacteria and cell debris are actively hunted by microglial cells. (D) A microglial cell emerging through a protrusion (short arrows) in the tissue is trying to engulf free bacteria (arrowheads) and a big phagosome/cell filled with bacteria (asterisk), by means of a large cell pseudopodium (long arrows). The dotted line marks a necrotic cell with intact nuclei (n). (E) At 12h postinfection, numerous cell corpses with intact nuclei (n), cell debris, and phagosome-like particles of different size (arrowheads) are present at the surface of the tissues. (F) Surface of control tissue at 12h. Magnifications: (A, C, F) 2000×; (B) 500×; (D) 5000×; (E) 7585×. Scale bars: (A, D, F) 25μm; (B) 100μm; (C) 10μm; (E) 5μm.")
SEM analysis of control and infected brain organotypic cultures. (A) Shows the thickness of an organotypic culture, with the surface covered cells forming a flat surface. (B) Activation of microglia by L. monocytogenes shows numerous round cells which emerge from the tissue. (C) 5h after infection, bacteria and cell debris are actively hunted by microglial cells. (D) A microglial cell emerging through a protrusion (short arrows) in the tissue is trying to engulf free bacteria (arrowheads) and a big phagosome/cell filled with bacteria (asterisk), by means of a large cell pseudopodium (long arrows). The dotted line marks a necrotic cell with intact nuclei (n). (E) At 12h postinfection, numerous cell corpses with intact nuclei (n), cell debris, and phagosome-like particles of different size (arrowheads) are present at the surface of the tissues. (F) Surface of control tissue at 12h. Magnifications: (A, C, F) 2000×; (B) 500×; (D) 5000×; (E) 7585×. Scale bars: (A, D, F) 25μm; (B) 100μm; (C) 10μm; (E) 5μm.
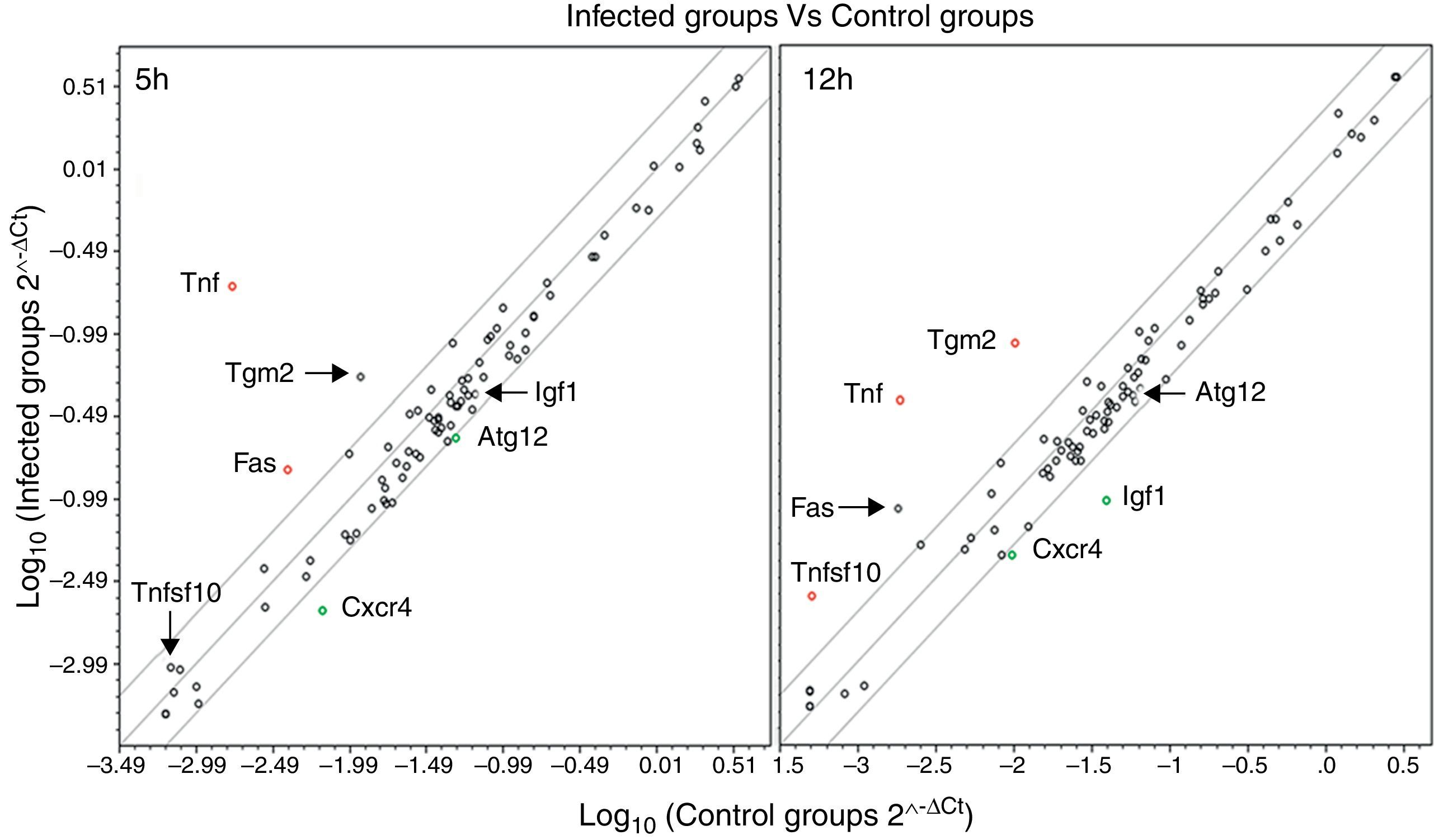
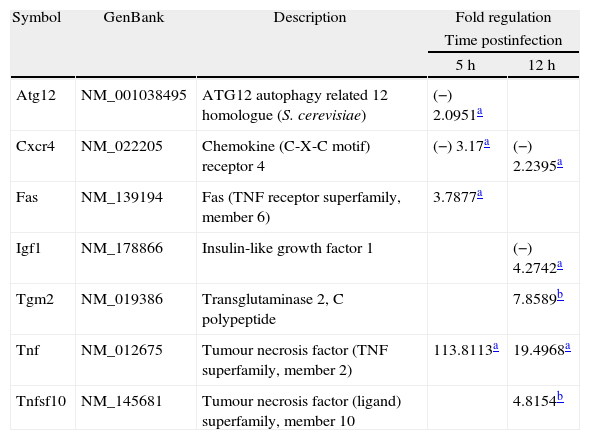
We used qPCR arrays to identify a large number of genes of the autophagy pathway that could be specifically modulated by the infection of L. monocytogenes in the brain. A list of all studied genes, their p-value, and fold regulation is shown in Tables S3 and S4. Our data show that all components of the autophagy machinery and other key regulators of autophagy are present and likely functional in newborn rat brains. The expression levels of 7 out of the 84 genes included in the autophagy q-PCR arrays used were clearly modified by the infection (Table 1 and Fig. 2). Tumour necrosis factor (TNF) was the most highly upregulated gene after infections. The expression of the Chemokine (C-X-C motif) receptor 4, and the insulin-like growth factor 1 was downregulated. Only one core autophagic gene (Atg12) was also downregulated by the infection whereas the others (Atg3, Atg4, Atg5, Atg7, Atg9, and Atg16) were not. Other important genes involved in autophagic vacuole formation present in the arrays were also not modulated by the infection, between them, Wipi1 and Ulk1. Important co-regulators of autophagy and apoptosis such as Bad, Bak1, Bax, Bcl2, Bid, Caspases 3 and 8, Tp53 and Tp73 were also not modulated by the infection.
Gene expression modulated by the infection.
| Symbol | GenBank | Description | Fold regulation | |
| Time postinfection | ||||
| 5h | 12h | |||
| Atg12 | NM_001038495 | ATG12 autophagy related 12 homologue (S. cerevisiae) | (−) 2.0951a | |
| Cxcr4 | NM_022205 | Chemokine (C-X-C motif) receptor 4 | (−) 3.17a | (−) 2.2395a |
| Fas | NM_139194 | Fas (TNF receptor superfamily, member 6) | 3.7877a | |
| Igf1 | NM_178866 | Insulin-like growth factor 1 | (−) 4.2742a | |
| Tgm2 | NM_019386 | Transglutaminase 2, C polypeptide | 7.8589b | |
| Tnf | NM_012675 | Tumour necrosis factor (TNF superfamily, member 2) | 113.8113a | 19.4968a |
| Tnfsf10 | NM_145681 | Tumour necrosis factor (ligand) superfamily, member 10 | 4.8154b | |
The results correspond to the mean values of three and four independent experiments (controls and infected tissues respectively), and are expressed as changes of infected tissues compared with uninfected controls. Fold-regulation represents fold-change results in a biologically meaningful way. A fold-change value greater than 1 indicates positive-or an upregulation, and the fold-regulation is equal to the fold-change. Fold-change values less than 1 indicate negative-or downregulation, and the fold-regulation is the negative inverse of the fold-change.
 altered expression (p<0.05 and are consistently up- or downregulated) in brains infected with L. monocytogenes compared with the expression in control tissues. Black dots outside the boundary lines indicate a fold regulation >2 (with p>0.05). Black dots inside the boundary lines indicate no change in regulation (<2-fold in either direction).")
Scatter plots comparing the expression of genes involved in the autophagy response in rat brains infected with L. monocytogenes and the expression of these genes in non-infected tissues at 5 and 12h. Each circle represents an individual gene. The central line indicates unchanged gene expression. The boundary lines indicate a twofold difference. Genes outside the boundary lines have twofold (>2, red dots or <2, green dots) altered expression (p<0.05 and are consistently up- or downregulated) in brains infected with L. monocytogenes compared with the expression in control tissues. Black dots outside the boundary lines indicate a fold regulation >2 (with p>0.05). Black dots inside the boundary lines indicate no change in regulation (<2-fold in either direction).
L. monocytogenes has been used as a model intracellular pathogen to study basic aspects of innate and acquired immunity. Complications from L. monocytogenes CNS infection include brain abscesses and inflammation where microglial activation plays a key role. Microglial cells are quickly activated in response to L. monocytogenes, producing a large array of cytotoxic factors which can affect the other cell types, leading to a neuroinflammation and abscess formation.13 Macroautophagy (here referred to as autophagy) has been recently proposed to be a component of the innate cellular immune response against intracellular pathogens.14 However, the interaction between pathogens and the autophagic pathway has mainly been studied using macrophage and epithelial cell lines, not microglia, and therefore information is lacking about autophagic events which may take place in the brain parenchyma. In the present study, we used q-PCR arrays containing 84 genes to analyse the expression profiles of autophagic genes in response to L. monocytogenes infection in the rat brain. Based on a previous study dealing with the kinetics of immune-related gene expression, the time points of 5h and 12h were selected because they provided good gene expression response with more than 20 genes modulated by the infections.13 Moreover, the intercellular spread of L. monocytogenes begins between 3 and 5h postinfection in vitro.15,16 Our results revealed that only a few genes of the core autophagic pathway were either up- or downregulated more than twofold over the course of the infections. In fact, only the expression of the core autophagy gene Atg12 was modulated by the infection. Surprisingly, the gene expression of other Atg genes (i.e. Atg3, Atg4, Atg5, Atg7, Atg9 and Atg16) remains unchanged with respect to the uninfected tissues. These results are in agreement with other studies reported elsewhere, where some pathogenic bacteria also manipulate autophagy regulation at the level of gene transcription. For example, Francisella tularensis, Yersinia enterocolitica and Burkholderia cenocepacia can downregulate the transcription of important Atg genes.17–19 Other microorganisms have evolved strategies to evade or subvert host autophagy to survive and establish a persistent infection.20,21
The expression of TNF and CXCR4 correlates well with our previous observations.13 This reinforces the robustness of the infection model and demonstrate that these cytokines play an active role during the response to L. monocytogenes infection in the brain. TNF plays an important role for pathogen control, limiting brain damage in a murine cerebral model of listeriosis.22 The inflammatory mediator CXCR4 was downregulated by the infection, at both 5 and 12h postinfection, whereas the insulin-like growth factor 1 was only at 12h. These genes act as co-regulators of the autophagy pathway.23,24
Interestingly, transglutaminase 2 (Tgm2) was consistently upregulated in all the infected tissues with respect to the controls at 12h (despite not statistically significant, Table S4). Tissue transglutaminase 2 belongs to a family of transglutaminase proteins that confers mechanical resistance from proteolysis and stabilises proteins. Tgm2 is also expressed in rat brain astrocytes in vitro, and is induced by the inflammation-associated cytokines interleukin-1beta and to a lesser extent by TNF-α.25 Importantly for our work, Akar and coworkers showed that tissue transglutaminase inhibits autophagy in pancreatic cancer cells.26 However, the role and the mechanisms that regulate Tgm2 expression in brain tissues remain elusive.
As judged from the modest increase in core autophagy gene expression in the 5–12h time course, a weak autophagic programme was activated in the rat brain after L. monocytogenes infection. Thus, identification of mechanisms or virulence factors exploiting autophagy may provide a new strategy for therapeutic intervention in infectious diseases. On the other hand, it has been hypothesised that basal levels of autophagy occur continuously inside of cells, and that an increase in autophagy can be stimulated by microbial infections.20 In this sense, there are many ways to study autophagy, from transmission scanning electron microscopy to immunofluorescence, and one limitation of the present study is that we used only real-time PCR to measure autophagic gene expression. However, our results offer an approach for capturing the whole picture of gene expression of core autophagy genes in the brain, and highlight the need to study context-specific host–pathogen interactions in brain infections, where autophagy seems to play a role in homeostasis, predominantly in a housekeeping process. Moreover, to date, few reports on the role of the autophagy pathway in the CNS have been published, and the results obtained also contribute to an understanding of the participation of microglia in the brain abscesses caused by L. monocytogenes and the genesis of brain injury associated with the pathogen.
The role of autophagy during L. monocytogenes infection in the brain is only just beginning to be appreciated. Increased knowledge about the autophagic pathway itself in the brain, in an organotypic context, or even in immortalised microglial cell lines, should be gained to understand the biology of neurotropic pathogens and their interaction with this degradative and immunologically relevant pathway. A better understanding of the participation of microglial cells in brain abscess formation and neuronal cell death opens up the possibility of manipulating microglial cells to reduce the impact of neuroinflammation. In addition, characterisation of the autophagic pathway in the brain awaits further experimentation which will contribute to our knowledge of the intracerebral immune response to L. monocytogenes and of the expanding functions of autophagy in innate and adaptive immune responses.
Ethical disclosuresProtection of human and animal subjects.
The authors declare that the procedures followed were in accordance with the regulations of the responsible Clinical Research Ethics Committee and in accordance with those of the World Medical Association and the Helsinki Declaration.
Confidentiality of Data.
The authors declare that no patient data appears in this article.
Right to privacy and informed consent.
The authors declare that no patient data appears in this article.
Conflict of interestThe authors have no conflict of interest to declare.
SRM holds a research contract from the Instituto de Formación e Investigación Marqués de Valdecilla (IFIMAV). JRV holds a Miguel Servet contract for Young Researchers from the Instituto de Salud Carlos III, Spain. The Instituto de Salud Carlos III (PS: CP08/100) and the Fondo de Investigaciones Sanitarias (FIS: PS09/00466) provide the financial support for this study and grants to JRV. EMV was supported by the Ministerio de Ciencia e Innovación (grants SAF07-61862 and SAF2011-25020). JMI was supported by the Spanish Ministerio de Economia y Competitividad (grant CGL2008-04559/BOS). We thank Verónica Inés Vargas and Beatriz Romero Presno for their technical assistance.
The following are the supplementary data to this article: