






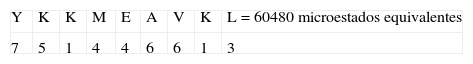
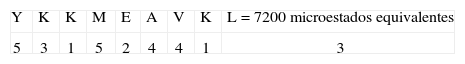
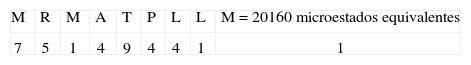
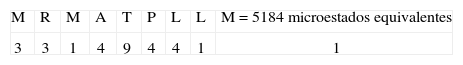
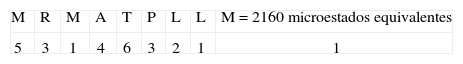
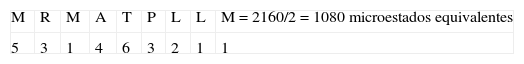
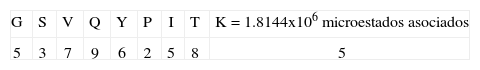
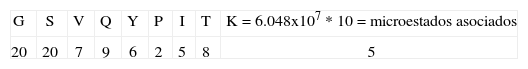

La presentación de péptidos antigénicos por las moléculas de histocompatibilidad a los receptores T es fundamental en la especificidad y la variabilidad de la defensa por parte del sistema inmune, por lo tanto un entendimiento físico y matemático de este fenómeno es de vital importancia para el diseño de vacunas.
Se calcularon valores de probabilidad, combinatoria y entropía de 100 secuencias específicas de péptidos teóricos, promiscuos, sintéticos y naturales que se unen a moléculas de HLA clase II y 61 péptidos que no se unen. Estos valores se usaron para diferenciar los péptidos seleccionados y desarrollar una caracterización física y matemática, objetiva y reproducible para el fenómeno de unión.
Se desarrolló una predicción teórica del conjunto apropiado de secuencias que debe tener un péptido para que sea presentado por la molécula de HLA clase II, acertando en todos los casos.
La teoría desarrollada facilita el proceso de selección
The antigenic peptide presentation by the histocompatibility molecules to the T receptors is fundamental in the specificity and variability of the host defense by the immune system, therefore a physical and mathematical understanding of this phenomenon is of vital importance for the design of vaccines.
Values of probability, combinatory and entropy from 100 specific sequences of theoretical, promiscuous, synthetic and natural peptides that bind to HLA class II molecules, as well as 61 peptides that do not bind were calculated. These values were used to differentiate the selected peptides and to develop an objective and reproducible physical and mathematical characterization of the binding phenomena.
A theoretical prediction of the appropriate group of sequences that a peptide should have so that it is presented by the HLA class II molecule was developed, hitting all the cases.
The developed theory facilitates the process of peptide selection required in the design of synthetic or natural vaccines.
La probabilidad es una medida predictiva de la posible ocurrencia de un evento, cuyo valor numérico se encuentra en el intervalo real [0, 1]. Kolmogorov definió la función de probabilidad como aquella que satisface tres axiomas elementales; la probabilidad de cualquier elemento del espacio muestral es mayor o igual a cero, la probabilidad de todo el espacio muestral es uno, y la probabilidad de la unión de eventos disyuntos es la suma de sus probabilidades(1-4). Esta teoría se usó para cuantificar las diferentes posibilidades de secuencias de péptidos teóricos y/o con capacidad o no de unión.
En el desarrollo de la Termodinámica y de la Mecánica Estadística, el concepto de entropía se ha ido reformulando y reinterpretando(5-8). La formulación enunciada por Boltzmann está dada en términos del número de microestados y se aplica, entre otros, a los sistemas con una distribución equiprobable(6, 7). En este trabajo se estudia la aparición de aminoácidos en secuencias de péptidos que podrían unirse o no al HLA de clase II, asumiendo que cada uno de ellos tiene la misma probabilidad de conformar el péptido; por ello, en analogía con los microestados de la mecánica estadística, la aplicación de la formulación de Boltzmann del concepto de entropía lleva a una medida del sistema que caracteriza la unión peptídica al HLA de clase II.
En un sistema de muchos cuerpos se definen los conceptos de macroestado y microestado. El macroestado es una configuración general del sistema que se puede manifestar mediante múltiples microestados posibles. Por ejemplo, dado un número de partículas en un volumen fijo existen múltiples configuraciones con densidad uniforme, la densidad uniforme es una característica del sistema que se puede expresar a través de diferentes microestados, que serían los diferentes arreglos posibles de organización de las partículas en el volumen(6, 7). En la presente investigación se usa una analogía para representar las secuencias de aminoácidos en términos de micro y macroestados (ver Definiciones)
El antígeno leucocitario humano, HLA, es un complejo de proteínas especializadas cuya función es la de presentar asociados a células los antígenos de los microorganismos para su reconocimiento por las células T. Las moléculas de HLA clase I presentan péptidos a las células T CD8+, mientras que las moléculas de HLA clase II presentan péptidos a las células T CD4+(9). Cada molécula de HLA está anclada a la célula mediante dominios transmembrana y citoplasmáticos y posee una hendidura extracelular de unión al péptido seguida de un par de dominios tipo inmunoglobulina(10).
En las moléculas de HLA clase II la hendidura de unión se presenta abierta en los dos extremos(11-14); debido a esto los péptidos presentados poseen un grado de variabilidad en su tamaño, comprendido entre 10 y 24 residuos; un tamaño de 13 a 16 aminoácidos ha sido observado con mayor frecuencia(15-18). Análisis de unión sugieren que únicamente una región central compuesta por nueve aminoácidos es esencial para la unión péptido-HLA clase II(19), estudios de dicha región han relacionado una alta frecuencia de aparición de un grupo de aminoácidos en una posición específica, posiciones de anclaje, con los denominados motivos de unión(20), construidos a partir de grupos seleccionados, por ejemplo, de librerías M13 de péptidos presentados.
El objeto de estudio de la presente investigación es un sistema de 20 aminoácidos que se organizan en 9 espacios de ocupación en la región central de péptidos antigénicos; y el propósito es construir una teoría general basada en las leyes de probabilidad, combinatoria y entropía, capaz de predecir la unión de péptidos a moléculas de HLA clase II.
DEFINICIONESMacroestados y microestados de las secuencias de aminoácidos (Definición del autor)Macroestado: secuencias de péptidos elegidos para diferenciar matemática y físicamente la capacidad de unión al HLA clase II, y aquellos que tengan el valor predicho para poder unirse. Por lo que existen dos clases de macroestados, los de unión y los de no unión.
Microestado: cualquier secuencia específica de nueve aminoácidos.
Tipo de secuencia: microestados que presentan el mismo valor en su combinatoria y su evaluación con los criterios definidos. Definimos 7 tipos de secuencia iniciales. Dos de estos son representativos del macroestado de no unión, mientras que cinco son péptidos representativos del macroestado de unión.
Probabilidad LaplacianaLa probabilidad de un tipo de secuencia A se define como el cociente entre la cantidad de microestados NA asociados a este tipo de secuencia y el total de posibles microestados N(1, 4).
Ley combinatoria para determinar la cantidad de microestados W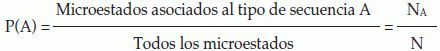
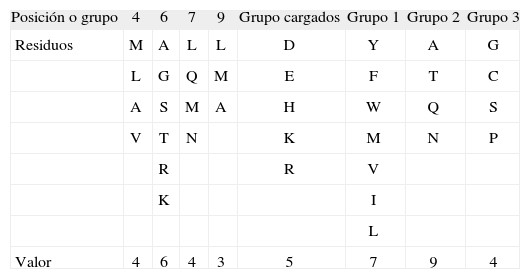
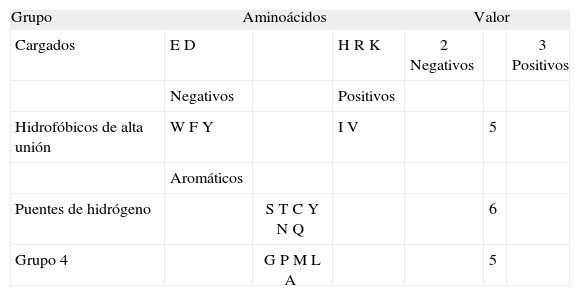
En la construcción del péptido, cada posición tiene asociada un número de posibles aminoácidos. La cantidad de posibles microestados asociados a un tipo de secuencia se calcula multiplicando los valores asociados a cada posición, lo cual depende del número de posibles aminoácidos asociados a cada lugar; esto está determinado por el grupo al que pertenezca y sus repeticiones en el tipo de secuencia(3, 4). Para las cuantificaciones específicas, ver tablas de criterios evaluadores y tablas de grupos de aminoácidos (Tablas I-IV).
Criterios evaluadores a partir de los cuales se calcula el número de microestados equivalentes para un tipo de secuencia específico
CRITERIO EVALUADOR
|
Criterios evaluadores a partir de los cuales se calcula el número de microestados equivalentes para un tipo de secuencia específico
CRITERIO EVALUADOR
|
En un sistema cuyos microestados son equiprobables la entropía está dada por:
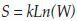
Donde k es igual a la constante de Boltzmann (1.38x10-23 (J/k)), W los posibles microestados y S el valor de la entropía(6, 7). Se eligió la formulación más simple para mayor sencillez de la teoría.
MATERIAL Y MÉTODOSEl objeto de estudio de la presente investigación es el sistema de 20 aminoácidos clasificados según características químicas, motivos y grupos, que pueden ser organizados en 9 posibles lugares de ocupación en la región central del HLA clase II. Los eventos posibles de posible ocupación son estudiados con las probabilidades de aparición de cada uno de los 20 aminoácidos y sus posibles formas de combinación en la región central asociadas o no a la unión. Teóricamente es casi improbable esperar que se repita un mismo aminoácido 9 veces seguidas en la naturaleza; mientras que evaluar péptidos no repetitivos que se unan a diferentes alelos de HLA, como los péptidos promiscuos, son más probables, evaluando así existencia o inexistencia de secuencias de péptidos y que se unen o no a la molécula presentadora.
Este trabajo cuantificará fundamentalmente la región central de unión(19). Inicialmente se define un espacio muestral, donde cada microestado es considerado como un posible evento. Se seleccionaron siete tipos de secuencias para el análisis: un péptido teórico que tiene el mismo aminoácido en todas sus posiciones; la región central del péptido de la Influenza Hemaglutinina, HA309-317, que se une a la mayoría de los alelos DR(21, 22); dos péptidos artificiales que contienen tres y cinco aminoácidos repetidos en las últimas posiciones(23); la región central de la cadena invariante del péptido asociada a la clase II, CLIP91-99(20); la región central de un péptido de unión con tres lisinas en las posiciones 2, 5 y 7(23); y por último un péptido que contiene aminoácidos diferentes en todas las posiciones de la región central y no se une al HLA clase II(23). El criterio de selección de éstos péptidos fue establecido con el fin de obtener un espectro completo de valores diferenciadores de probabilidad combinatoria y entropía de secuencias que se unen de las que no, para caracterizarlos matemática y físicamente. El péptido teórico se escogió para ser contrastado con las combinatorias de aminoácidos naturales y artificiales, los péptidos promiscuos se escogieron por su capacidad de unión a diferentes alelos, los péptidos artificiales y el péptido con tres lisinas, K, para caracterizar la importancia de las repeticiones de aminoácidos respecto a la unión, el último péptido que tiene todos los aminoácidos diferentes y no presenta unión, se escogió para compararlo con las secuencias de unión y con el péptido teórico donde todos los aminoácidos son repetidos, para diferenciar unión de no unión con repeticiones o no de aminoácidos.
Se calculó la probabilidad laplaciana del tipo de secuencia en el que todos los aminoácidos son iguales y se comparó con el valor obtenido para el tipo de secuencia en el cual las tres primeras posiciones están determinadas por un aminoácido, tomando como ejemplo la secuencia de la librería M13 de péptidos presentados(21) donde las tres primeras posiciones son tres triptófanos (W).
Posteriormente se evaluó la probabilidad laplaciana de cada uno de los restantes tipos de secuencia, aplicando la ley combinatoria (ver definiciones), para calcular los microestados equivalentes a partir de la cantidad de aminoácidos que tendrían la posibilidad de ocupar cada posición. El número de aminoácidos asociados a cada posición depende del valor del grupo al que pertenezca y sus repeticiones en el tipo de secuencia. Para estos cálculos se realizaron cuatro pasos; en el primero se definieron 8 grupos de aminoácidos: los 7 aminoácidos hidrofóbicos característicos de unión en la posición 1(23); aminoácidos cargados; los aminoácidos A, T, Q, N; un grupo con los aminoácidos G, C, S, P; y finalmente motivos reconocidos para posiciones 4, 6, 7 y 9 según resultados experimentales(21) (Tabla I). El segundo paso consiste en evaluar diez criterios basados en las tres leyes teóricas y en observaciones experimentales (Tabla II). En el paso 3 se evalúan los péptidos que en el paso 1-2 presentaron el macroestado asociado a la unión, definiendo 4 grupos de aminoácidos: aminoácidos cargados, aminoácidos que hacen puentes de hidrógeno, 3 aminoácidos hidrofóbicos de baja unión(23) junto con la glicina y la prolina, y 5 aminoácidos hidrofóbicos característicos de alta unión en la posición 1(23) (Tabla III). El cuarto paso consiste en evaluar diez criterios basados en las tres leyes teóricas y en observaciones experimentales (Tabla IV).
Los pasos tres y cuatro, aplicados a los macroestados de unión resultados de la aplicación de los pasos 1 y 2, se usaron para refinar los cálculos.
El procedimiento de cálculo para los pasos uno y tres es asignar a cada posición un número según el aminoácido encontrado en cada lugar dependiendo del conjunto en el que se encuentre. Se realiza el mismo procedimiento con cada aminoácido que aparece consecutivamente, considerando además la disminución de sus posibilidades en la medida que van apareciendo aminoácidos de un mismo grupo y las repeticiones de un mismo aminoácido. Posteriormente, para calcular la probabilidad laplaciana de un tipo de secuencia (ver definiciones), se multiplican dichos valores hallando el número de microestados asociados al tipo de secuencia (Tablas V y VI). La metodología desarrollada es ejemplificada para cuatro secuencias específicas (Anexo 1).
Pasos 1-2: Cálculos de probabilidad y número de microestados
| Secuencia | Probabilidad del tipo de secuencia | Número de microestados |
| Péptido teórico: GGGGGGGGG* | 20/209=3.9x10-11 | 20 |
| CLIP91-99: MRMATPLLM | 5184/209=1,01 x10-8 | 5184 |
| Péptido artificial: YRAMAAAAA | 6480/209=1,26 x10-8 | 6480 |
| Péptido artificial: YRAMSTAAA | 7776/209=1,51x10-8 | 7776 |
| YKKMEAVKL** | 60480/209=1,18 x10-7 | 60480 |
| HA309-317: YVKQNTLKL | 362880/209=7,08 x10-7 | 362880 |
| Aminoácidos diferentes en todas las posiciones: | ||
| GSVQYPITK | 604'800.000/209=1,1 x10-3 | 604'800.000 |
Los valores para esta secuencia son calculados teóricamente, ver Anexo 1, y no se aplicaron las reglas del segundo paso
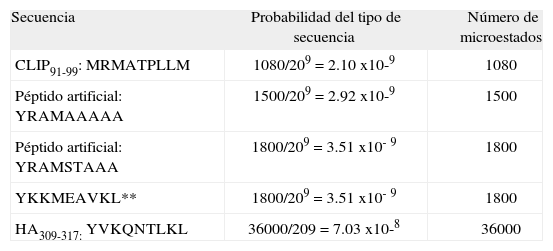
Pasos 3-4: Cálculos de probabilidad y número de microestados
| Secuencia | Probabilidad del tipo de secuencia | Número de microestados |
| CLIP91-99: MRMATPLLM | 1080/209=2.10 x10-9 | 1080 |
| Péptido artificial: YRAMAAAAA | 1500/209=2.92 x10-9 | 1500 |
| Péptido artificial: YRAMSTAAA | 1800/209=3.51 x10-9 | 1800 |
| YKKMEAVKL** | 1800/209=3.51 x10-9 | 1800 |
| HA309-317: YVKQNTLKL | 36000/209=7.03 x10-8 | 36000 |
Cálculos de microestados y entropía
Este tipo de secuencia tiene la característica de tener en todas sus posiciones el mismo aminoácido, por esto a la primera posición se le asigna un valor de 20 pues puede aparecer cualquiera de los 20 aminoácidos esenciales, las siguientes posiciones tienen un valor de 1 pues se repite el mismo aminoácido de la primera posición.
El valor de entropía asociado a este tipo de secuencia tiene un valor de:
2. Tipo de secuencia: YKKMEAVKL Paso uno: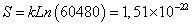
Este tipo de secuencia está asociado a un péptido de unión (22), la primera posición Y tiene un valor de 7 pues pertenece al grupo 1 (Tabla I) en la posición dos a K se le otorga un valor de 5, dado que está dentro del grupo de cargados, la posición 3 tiene un valor de uno pues el aminoácido ya había aparecido, a M le corresponde 4 porque pertenece al grupo de motivos en esa posición, a E le corresponde un valor de 4 porque está en el grupo de aminoácidos cargados y el valor disminuye pues en la posición dos apareció un aminoácido del mismo grupo. A tiene valor 6, pues pertenece al grupo de motivos en esa posición; a V le corresponde un valor de 6 porque está en el grupo 1 y el valor disminuye pues en la posición uno apareció un aminoácido del mismo grupo. La posición 8 tiene un valor de 1, pues el aminoácido ya había aparecido; finalmente a L le corresponde 3 pues pertenece al grupo de motivos en esa posición.
Paso dos:Para este tipo de secuencia no se cumple ningún criterio, por lo tanto se mantiene el valor calculado en el primer paso. Posteriormente se calcula el valor de entropía asociado al tipo de secuencia por medio de la ecuación 2:
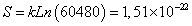
Dado que el tipo de secuencia asociado está dentro de los rangos de unión, se realiza el tercer paso:
Paso tres:Este tipo de secuencia está asociado a un péptido de unión (22). A la primera posición Y se le otorga un valor de 5, pues pertenece al grupo de 5 aminoácidos hidrofóbicos de alta unión (Tabla III). En la posición dos a K se le otorga un valor de 3, dado que está dentro del grupo de cargados positivos; a la posición 3 se le otorga un valor de uno, pues el aminoácido ya había aparecido en la posición 2. A la posición 4 (M) se le da un valor de 5, pues pertenece a los aminoácidos del grupo 4. En la posición cinco (E) se le otorga un valor de 2, ya que está dentro del grupo de cargados negativos. A A le corresponde 4 porque pertenece al grupo 4, y el valor decrece debido a que en la posición 4 apareció un aminoácido del mismo grupo. A V le corresponde 4 porque pertenece al grupo de hidrofóbicos de alta unión y el valor decrece por la aparición de otro aminoácido del mismo grupo en la posición 1. A la posición 8 (K) se le otorga un valor de uno, pues el aminoácido ya había aparecido en la posición 2. Finalmente, en la última posición 9 se le da un valor de tres, porque pertenece al grupo 4 y el valor decrece debido a que en la posición 4 y 6 aparecieron aminoácidos del mismo grupo.
Posteriormente se calcula el valor de entropía asociado al tipo de secuencia por medio de la ecuación 2:
Paso cuatro: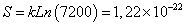
Para este tipo de secuencia no se cumple ningún criterio, por lo tanto se mantiene el valor calculado en el tercer paso.
3. Tipo de secuencia: MRMATPLLMPaso uno:Este tipo de secuencia está asociado al péptido CLIP. La primera posición M tiene un valor de 7 pues pertenece al grupo 1 (Tabla I); en la posición dos a R se le otorga un valor de 5, dado que está dentro del grupo de cargados; a M le corresponde un valor de 1, pues el aminoácido ya había aparecido. La A siguiente tiene un valor de 4 porque pertenece al grupo de motivos en esa posición; el siguiente aminoácido tiene un valor de 9 pues pertenece al grupo 2 y no hay motivo para esa posición; la P tiene un valor de 4 pues pertenece al grupo 3 y no es motivo para esa posición. La posición 7 tiene un valor de 4, pues pertenece al grupo de motivos para esa posición; las dos últimas posiciones tienen un valor de 1, pues estos aminoácidos ya habían aparecido.
Paso dos:Para este tipo de secuencia se cumple el criterio número 1, pues en la posición uno tenemos M un aminoácido hidrofóbico de unión, y además en la segunda posición aparece una R; entonces estas dos posiciones toman un valor de 3:
Posteriormente se calcula el valor de entropía asociado al tipo de secuencia por medio de la ecuación 2:
Paso tres: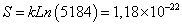
A la primera posición M se le otorga un valor de 5, pues pertenece al grupo 4 (Tabla III); en la posición dos a R se le otorga un valor de 3, dado que está dentro del grupo de cargados positivos. Luego aparece nuevamente M, y se le otorga un valor de 1; a la posición 4 le corresponde 4 ya que pertenece al grupo 4 y el valor decrece debido a que en la posición uno apareció un aminoácido del mismo grupo; la T siguiente tiene un valor de 6 pues pertenece al grupo que hacen puentes de hidrógeno. La P siguiente tiene un valor de 3, pues pertenece al grupo 4 y el valor decrece; la siguiente posición tiene un valor de 2 pues pertenece al grupo 4 y el valor decrece; las dos últimas posiciones tienen un valor de 1, pues están repetidos.
Paso cuatro:Para este tipo de secuencia se cumple el criterio número 1, pues en la posición uno, tres, siete, ocho y nueve tenemos aminoácidos hidrofóbicos de unión, es decir, la secuencia tiene un total de cinco aminoácidos hidrofóbicos de unión, entonces el número de microestados se divide entre dos.
Posteriormente se calcula el valor de entropía asociado al tipo de secuencia por medio de la ecuación 2:
4. Tipo de secuencia: GSVQYPITKPaso uno: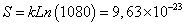
Este tipo de secuencia tiene la característica de tener en todas sus posiciones diferentes aminoácidos. La primera posición tiene un valor de 4, pues pertenece a un aminoácido del grupo tres (Tabla I); la siguiente posición toma un valor de tres, pues pertenece a este mismo grupo y el valor decrece, y la siguiente posición toma un valor de 7 pues pertenece al grupo 1. La siguiente posición toma un valor de 9 pues pertenece al grupo 2; la posición 5 toma un valor de 6 pues pertenece al grupo 1 y el valor decrece debido a la aparición de un aminoácido del mismo grupo en a posición 3. La siguiente posición toma un valor de 2, pues pertenece a este mismo grupo y el valor decrece; la posición 7 toma un valor de 5, pues pertenece al grupo 1 y el valor decrece; la posición 8 toma un valor de 8, pues pertenece al grupo 2 y el valor decrece por la anterior aparición en la posición 4 de un aminoácido del mismo grupo. Finalmente, la posición 9 toma un valor de 5, pues es un aminoácido cargado.
Paso dos:Para este tipo de secuencia se cumple el criterio dos, pues no presenta uno de los 7 aminoácidos hidrofóbicos característicos de unión en la posición 1, entonces esta posición toma un valor de 20. Además, en la segunda posición no presenta una R, entonces también toma un valor de 20; por otra parte se cumple el criterio 8, pues no presenta uno de los 7 aminoácidos hidrofóbicos característicos de unión en la posición 1, en la segunda posición no presenta una R, y la secuencia tiene menos de 4 repeticiones, entonces el número de microestados se multiplica por 10:
El valor de entropía asociado a este tipo de secuencia tiene un valor de:
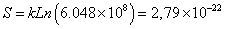
Todas las posibles combinaciones de aminoácidos generan secuencias específicas que pueden ser asociadas al fenómeno de unión, o no. Si se considera que en cada lugar de ocupación cada aminoácido tiene la misma probabilidad de aparición, y que basados en datos de la biología molecular damos valores diferentes para aminoácidos característicos de la unión en ciertas posiciones, y se evalúa cada secuencia específica como un microestado (ver definiciones), es posible caracterizar matemáticamente con la ley de entropía todas las posibles asociaciones de aminoácidos y relacionarlas con unión al HLA o no.
Partiendo del número de microestados posibles se realizaron los cálculos de entropía para cada tipo de secuencia aplicando la ley de Boltzmann (Ecuación 2), buscando caracterizaciones matemáticas estrictas para cualquier cadena nonamérica (Tablas VII y VIII).
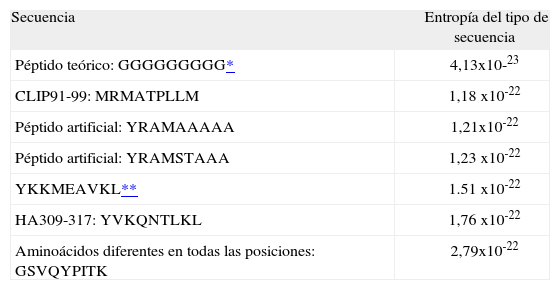
Pasos 1 y 2: Cálculos de entropía
| Secuencia | Entropía del tipo de secuencia |
| Péptido teórico: GGGGGGGGG* | 4,13x10-23 |
| CLIP91-99: MRMATPLLM | 1,18 x10-22 |
| Péptido artificial: YRAMAAAAA | 1,21x10-22 |
| Péptido artificial: YRAMSTAAA | 1,23 x10-22 |
| YKKMEAVKL** | 1.51 x10-22 |
| HA309-317: YVKQNTLKL | 1,76 x10-22 |
| Aminoácidos diferentes en todas las posiciones: GSVQYPITK | 2,79x10-22 |
Los valores para esta secuencia son calculados teóricamente (Ver Anexo 1) y no se aplicaron las reglas del segundo paso.
A partir del rango de microestados y de entropía asociado a la unión, se realiza la predicción teórica del número de microestados equivalentes y la configuración particular para los péptidos de unión a la molécula de HLA clase II, a partir de la ecuación de Boltzmann (Ecuación 2). Despejando W, que es el número de microestados asociados a cada tipo de secuencia, física y matemáticamente se deduce cuales son las secuencias de aminoácidos que se unen (Ecuación 3).
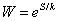
Posteriormente, a partir de la ecuación 2, se despejó el valor de S/k (Ecuación 4).
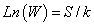
Con la proporción S/k se predicen cuales son los valores de los péptidos asociados al macroestado unión y cuales son los asociados al macroestado de no unión (Tablas IX y X).
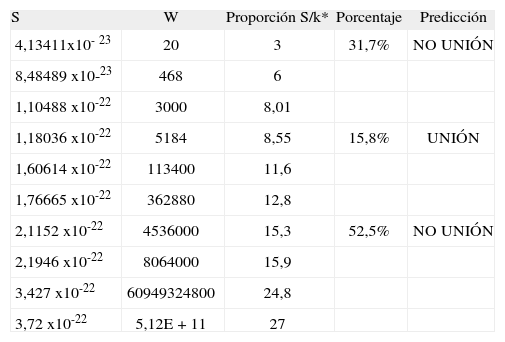
Predicción teórica del número de microestados W asociados a un valor particular de entropía S el valor de la proporción S/k asociado, y su porcentaje. Donde k es igual a la constante de Boltzmann con un valor de 1.38x10-23 (J/k), paso 1-2
| S | W | Proporción S/k* | Porcentaje | Predicción |
| 4,13411x10-23 | 20 | 3 | 31,7% | NO UNIÓN |
| 8,48489 x10-23 | 468 | 6 | ||
| 1,10488 x10-22 | 3000 | 8,01 | ||
| 1,18036 x10-22 | 5184 | 8,55 | 15,8% | UNIÓN |
| 1,60614 x10-22 | 113400 | 11,6 | ||
| 1,76665 x10-22 | 362880 | 12,8 | ||
| 2,1152 x10-22 | 4536000 | 15,3 | 52,5% | NO UNIÓN |
| 2,1946 x10-22 | 8064000 | 15,9 | ||
| 3,427 x10-22 | 60949324800 | 24,8 | ||
| 3,72 x10-22 | 5,12E+11 | 27 |
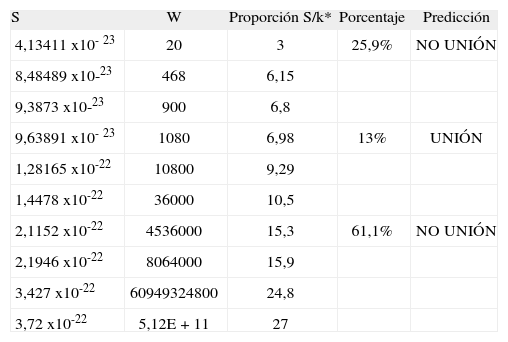
Predicción teórica del número de microestados W asociados a un valor particular de entropía S y el valor de la proporción S/k asociado, y su porcentaje. Donde k es igual a la constante de Boltzmann con un valor de 1.38x10-23 (J/k), paso 3-4
| S | W | Proporción S/k* | Porcentaje | Predicción |
| 4,13411 x10-23 | 20 | 3 | 25,9% | NO UNIÓN |
| 8,48489 x10-23 | 468 | 6,15 | ||
| 9,3873 x10-23 | 900 | 6,8 | ||
| 9,63891 x10-23 | 1080 | 6,98 | 13% | UNIÓN |
| 1,28165 x10-22 | 10800 | 9,29 | ||
| 1,4478 x10-22 | 36000 | 10,5 | ||
| 2,1152 x10-22 | 4536000 | 15,3 | 61,1% | NO UNIÓN |
| 2,1946 x10-22 | 8064000 | 15,9 | ||
| 3,427 x10-22 | 60949324800 | 24,8 | ||
| 3,72 x10-22 | 5,12E+11 | 27 |
A continuación se calcularon los valores de probabilidad, microestados y entropía para 12 regiones centrales de péptidos probados experimentalmente de unión(23); todas las cadenas nonámeras sobrelapadas de 12 secuencias de 13 aminoácidos de largo de no unión, probadas experimentalmente(23) (Tabla XI); y 83 péptidos de la página Web: www.immuneepitope. org(24) (Anexo 2). Para éstos últimos los cálculos se efectuaron a todas las posibles cadenas nonámeras sobrelapadas (Tabla XII). Fueron utilizados un total de 161 péptidos, 100 de unión y 61 de no unión, contrastando los resultados con la predicción teórica.
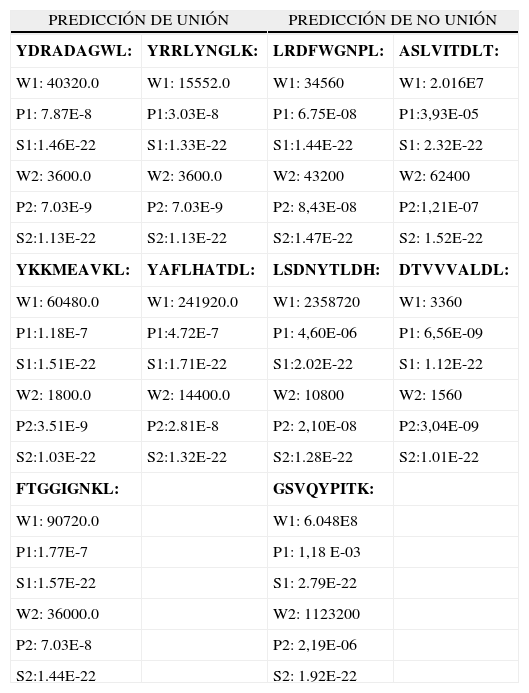
Cálculos realizados para regiones centrales de péptidos probados experimentalmente(23). Resultados escogidos de 24 secuencias. W1, P1, S1, son los valores de microestados, probabilidad y entropía para el paso 1-2. W2, P2, S2, son los valores de microestados, probabilidad y entropía para el paso 3-4.
| PREDICCIÓN DE UNIÓN | PREDICCIÓN DE NO UNIÓN | ||
| YDRADAGWL: | YRRLYNGLK: | LRDFWGNPL: | ASLVITDLT: |
| W1: 40320.0 | W1: 15552.0 | W1: 34560 | W1: 2.016E7 |
| P1: 7.87E-8 | P1:3.03E-8 | P1: 6.75E-08 | P1:3,93E-05 |
| S1:1.46E-22 | S1:1.33E-22 | S1:1.44E-22 | S1: 2.32E-22 |
| W2: 3600.0 | W2: 3600.0 | W2: 43200 | W2: 62400 |
| P2: 7.03E-9 | P2: 7.03E-9 | P2: 8,43E-08 | P2:1,21E-07 |
| S2:1.13E-22 | S2:1.13E-22 | S2:1.47E-22 | S2: 1.52E-22 |
| YKKMEAVKL: | YAFLHATDL: | LSDNYTLDH: | DTVVVALDL: |
| W1: 60480.0 | W1: 241920.0 | W1: 2358720 | W1: 3360 |
| P1:1.18E-7 | P1:4.72E-7 | P1: 4,60E-06 | P1: 6,56E-09 |
| S1:1.51E-22 | S1:1.71E-22 | S1:2.02E-22 | S1: 1.12E-22 |
| W2: 1800.0 | W2: 14400.0 | W2: 10800 | W2: 1560 |
| P2:3.51E-9 | P2:2.81E-8 | P2: 2,10E-08 | P2:3,04E-09 |
| S2:1.03E-22 | S2:1.32E-22 | S2:1.28E-22 | S2:1.01E-22 |
| FTGGIGNKL: | GSVQYPITK: | ||
| W1: 90720.0 | W1: 6.048E8 | ||
| P1:1.77E-7 | P1: 1,18 E-03 | ||
| S1:1.57E-22 | S1: 2.79E-22 | ||
| W2: 36000.0 | W2: 1123200 | ||
| P2: 7.03E-8 | P2: 2,19E-06 | ||
| S2:1.44E-22 | S2: 1.92E-22 | ||
Péptidos utilizados de la página web: http://www.immuneepitope.org(24)
| 1. | DFEFEQMFTDAMG | 29. | FNNIPSRYNLYDKMLELDD | 57. | QFSLWRRPVVTAHIE |
| 2. | IIGYVIGTQQATPGP | 30. | DPIELNATLSAVA | 58. | RHNWVNHAVPLAMKLI |
| 3. | GREIIYPNASLLIQN | 31. | PRYISLIPVNVVAD | 59. | EKVPVSEVMGTTLAEMSTPEAT |
| 4. | LQLSNGNRTLTLFNV | 32. | PLNTSYRSGENLNLS | 60. | IYRRRLMKQDFSVPQLPHS |
| 5. | TLTLFNVTRNDTASY | 33. | YTNQNINISMERNLMKHGFH | 61. | KRCLLHLAVIGALLAVGATKV |
| 6. | TVSAELPKPSISSNN | 34. | AYGSFVRTVSL | 62. | PGPVTAQVVLQAAIPLTSCGSS |
| 7. | KVKILPKDRWTQHTTTGG | 35. | LTNQNINIDQEFNLMKHGFH | 63. | QNILLSNAPLGPQFP |
| 8. | YSWRINGIPQQHTQV | 36. | FAYGSFVRT | 64. | HQAISPRTLNSPAIF |
| 9. | LQLSNDNRTLTLLSV | 37. | QRLLLTASLLTFWNP | 65. | KILEPFRKYTAFTIP |
| 10. | QEIDPLSYNYIPVNSN | 38. | ITVSASGTSPGLSAG | 66. | FAKIKNTHTNGVRLL |
| 11. | LSYNYIPVN | 39. | ADLVPTATLLDTY | 67. | GFLGFLATAGSAMGA |
| 12. | SSVFNVVNSSIGLIM | 40. | FAYGSFVRTVSLPGADE | 68. | HGIRPVVSTQLLL |
| 13. | VVFPASFFIKLPIILA | 41. | AYGSFVRTVSLPGADE | 69. | ILQRLSATLQRIREV |
| 14. | FATCFLIPLTSQFFLP | 42. | TFWNPPTTAKLTIES | 70. | IRPVVSTQLLL |
| 15. | RVYQEPQVSPPQRAET | 43. | EFAYGSFVRTVSLPGADE | 71. | ITTEQEIQFQQSKNS |
| 16. | IVAVHVASGFIEAEV | 44. | ALFNRLLDDLGF | 72. | PKISFEPIPIHYCAPAGFAI |
| 17. | SAMGAASLTLTAQSR | 45. | YGSFVRTVSLPGADE | 73. | VWGIKQLQARVLAVERYLKD |
| 18. | THGIRPVVSTQLLL | 46. | EFAYGSFVRTVSL | 74. | ENPVVHFFANIVTPRTP |
| 19. | TIVLMAVHCMNFKRR | 47. | VLLLVHNLPQHLFGY | 75. | IPQEWKPAITVKVLPA |
| 20. | VILLRIVIYIVQMLA | 48. | FDLEMLGDVESPS | 76. | KSKYKLATSVLAGLL |
| 21. | VQCTHGIRPVVSTQLLL | 49. | NNSIVKSITVSASGT | 77. | KTVTAMDVVYALKRQ |
| 22. | VSTVQCTHGIRPVVSTQL | 50. | TVGIMIGVLVGVALI | 78. | LTSQFFLPALPVFTWL |
| 23. | WDFISTPPLVRLVFN | 51. | SWYKGERVDGNRQII | 79. | LWWSTMYLTHHYFVDL |
| 24. | WLSTYAVRITWYSKN | 52. | KVKILPRDRWTQHTTTGG | 80. | VKNVIGPFMKAVCVE |
| 25. | WRRDNRRGLRMAKQN | 53. | TISPSYTYYRPGVNL | 81. | SSIIFGAFPSLHSGCC |
| 26. | LIRILQRALFMHFRG | 54. | YLWWVNNQSLPVSPR | 82. | LAAIIFLFGPPTALRS |
| 27. | FNNIPSRYNLYDKMLPLDD | 55. | DTGFYTLHVIKSDLV | 83. | THHYFVDLIGGAMLSL |
| 28. | NNIPSRYNLYDKMLDLDDL | 56. | LRDNIQGITKPAIRR |
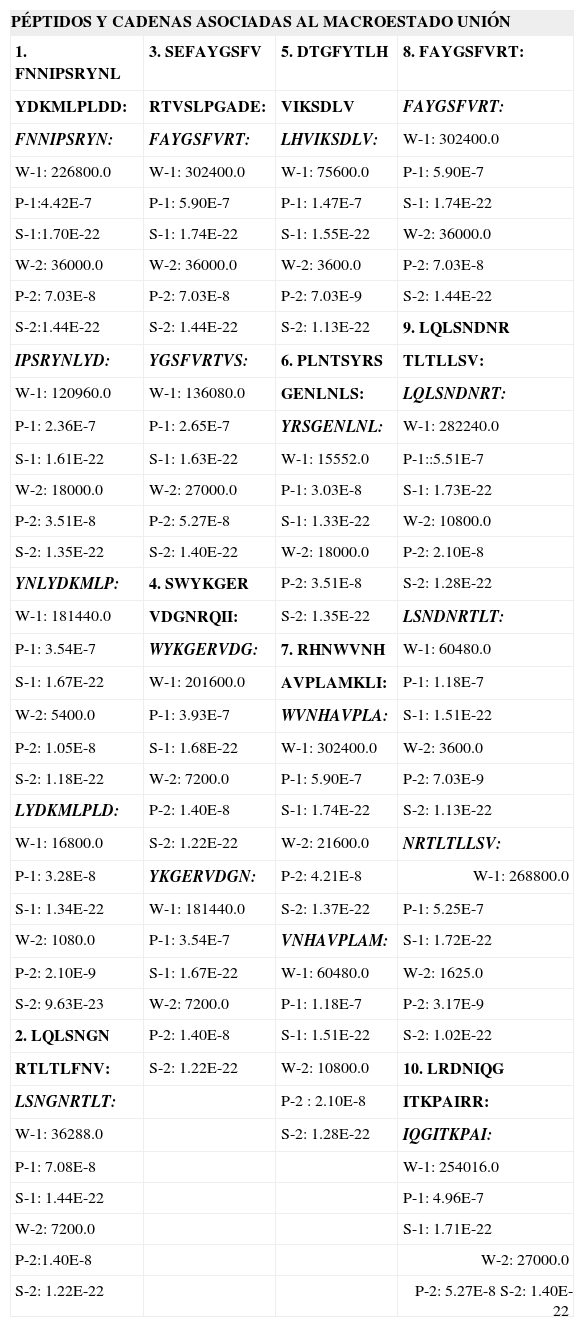
Cálculos realizados para regiones centrales predichas de unión para 10 péptidos de la página Web: www.immuneepitope.org (24). Resultados escogidos de 83 secuencias. W1, P1, S1, son los valores de microestados, probabilidad y entropía para el paso 1-2. W2, P2, S2, son los valores de microestados, probabilidad y entropía para el paso 3-4.
| PÉPTIDOS Y CADENAS ASOCIADAS AL MACROESTADO UNIÓN | |||
| 1. FNNIPSRYNL | 3. SEFAYGSFV | 5. DTGFYTLH | 8. FAYGSFVRT: |
| YDKMLPLDD: | RTVSLPGADE: | VIKSDLV | FAYGSFVRT: |
| FNNIPSRYN: | FAYGSFVRT: | LHVIKSDLV: | W-1: 302400.0 |
| W-1: 226800.0 | W-1: 302400.0 | W-1: 75600.0 | P-1: 5.90E-7 |
| P-1:4.42E-7 | P-1: 5.90E-7 | P-1: 1.47E-7 | S-1: 1.74E-22 |
| S-1:1.70E-22 | S-1: 1.74E-22 | S-1: 1.55E-22 | W-2: 36000.0 |
| W-2: 36000.0 | W-2: 36000.0 | W-2: 3600.0 | P-2: 7.03E-8 |
| P-2: 7.03E-8 | P-2: 7.03E-8 | P-2: 7.03E-9 | S-2: 1.44E-22 |
| S-2:1.44E-22 | S-2: 1.44E-22 | S-2: 1.13E-22 | 9. LQLSNDNR |
| IPSRYNLYD: | YGSFVRTVS: | 6. PLNTSYRS | TLTLLSV: |
| W-1: 120960.0 | W-1: 136080.0 | GENLNLS: | LQLSNDNRT: |
| P-1: 2.36E-7 | P-1: 2.65E-7 | YRSGENLNL: | W-1: 282240.0 |
| S-1: 1.61E-22 | S-1: 1.63E-22 | W-1: 15552.0 | P-1::5.51E-7 |
| W-2: 18000.0 | W-2: 27000.0 | P-1: 3.03E-8 | S-1: 1.73E-22 |
| P-2: 3.51E-8 | P-2: 5.27E-8 | S-1: 1.33E-22 | W-2: 10800.0 |
| S-2: 1.35E-22 | S-2: 1.40E-22 | W-2: 18000.0 | P-2: 2.10E-8 |
| YNLYDKMLP: | 4. SWYKGER | P-2: 3.51E-8 | S-2: 1.28E-22 |
| W-1: 181440.0 | VDGNRQII: | S-2: 1.35E-22 | LSNDNRTLT: |
| P-1: 3.54E-7 | WYKGERVDG: | 7. RHNWVNH | W-1: 60480.0 |
| S-1: 1.67E-22 | W-1: 201600.0 | AVPLAMKLI: | P-1: 1.18E-7 |
| W-2: 5400.0 | P-1: 3.93E-7 | WVNHAVPLA: | S-1: 1.51E-22 |
| P-2: 1.05E-8 | S-1: 1.68E-22 | W-1: 302400.0 | W-2: 3600.0 |
| S-2: 1.18E-22 | W-2: 7200.0 | P-1: 5.90E-7 | P-2: 7.03E-9 |
| LYDKMLPLD: | P-2: 1.40E-8 | S-1: 1.74E-22 | S-2: 1.13E-22 |
| W-1: 16800.0 | S-2: 1.22E-22 | W-2: 21600.0 | NRTLTLLSV: |
| P-1: 3.28E-8 | YKGERVDGN: | P-2: 4.21E-8 | W-1: 268800.0 |
| S-1: 1.34E-22 | W-1: 181440.0 | S-2: 1.37E-22 | P-1: 5.25E-7 |
| W-2: 1080.0 | P-1: 3.54E-7 | VNHAVPLAM: | S-1: 1.72E-22 |
| P-2: 2.10E-9 | S-1: 1.67E-22 | W-1: 60480.0 | W-2: 1625.0 |
| S-2: 9.63E-23 | W-2: 7200.0 | P-1: 1.18E-7 | P-2: 3.17E-9 |
| 2. LQLSNGN | P-2: 1.40E-8 | S-1: 1.51E-22 | S-2: 1.02E-22 |
| RTLTLFNV: | S-2: 1.22E-22 | W-2: 10800.0 | 10. LRDNIQG |
| LSNGNRTLT: | P-2 : 2.10E-8 | ITKPAIRR: | |
| W-1: 36288.0 | S-2: 1.28E-22 | IQGITKPAI: | |
| P-1: 7.08E-8 | W-1: 254016.0 | ||
| S-1: 1.44E-22 | P-1: 4.96E-7 | ||
| W-2: 7200.0 | S-1: 1.71E-22 | ||
| P-2:1.40E-8 | W-2: 27000.0 | ||
| S-2: 1.22E-22 | P-2: 5.27E-8 S-2: 1.40E-22 | ||
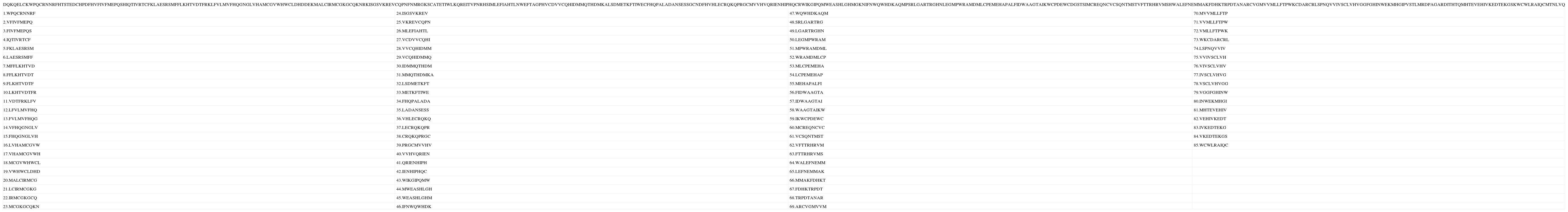
Finalmente, por medio de un algoritmo se generó una secuencia matemática de 500 aminoácidos, encontrando todas las cadenas nonámeras sobrelapadas para calcular sus valores de probabilidad, combinatoria, y entropía (ver Anexo 3 y Tabla XIII), y se estudiaron sus implicaciones.
Secuencia teórica generada, y péptidos de esta secuencia predichos como de unión
| DQKQELCKWPQCRNNRFHTSTEDCHPDFHVFIVFMEPQSHIQTIVRTCFKLAESRSMFFLKHTVDTFRKLFVLMVFHQGNGLVHAMCGVWHWCLDHDDEKMALCIRMCGKGCQKNRKISGSVKREVCQPNFNMRGKSCATETIWLKQREITVPNRHSIMLEFIAHTLNWEFTAGPHVCDVVCQHIDMMQTHDMKALSDMETKFTIWECFHQPALADANSESSGCNDFHVHLECRQKQPRGCMVVHVQRIENHIPHQCRWIKGIPQMWEASHLGHMGKNIFNWQWHDKAQMPSRLGARTRGHNLEGMPWRAMDMLCPEMEHAPALFIDWAAGTAIKWCPDEWCDGSTSIMCREQNCVCSQNTMSTVFTTRHRVMSHWALEFNEMMAKFDHKTRPDTANARCVGMVVMLLFTPWKCDARCRLSPNQVVIVSCLVHVGGFGHINWEKMHGIPVSTLMRDPAGARDITHTQMHTEVEHIVKEDTEKGSKWCWLRAIQCMTNLVQ | |||
| 1.WPQCRNNRF | 24.ISGSVKREV | 47.WQWHDKAQM | 70.MVVMLLFTP |
| 2.VFIVFMEPQ | 25.VKREVCQPN | 48.SRLGARTRG | 71.VVMLLFTPW |
| 3.FIVFMEPQS | 26.MLEFIAHTL | 49.LGARTRGHN | 72.VMLLFTPWK |
| 4.IQTIVRTCF | 27.VCDVVCQHI | 50.LEGMPWRAM | 73.WKCDARCRL |
| 5.FKLAESRSM | 28.VVCQHIDMM | 51.MPWRAMDML | 74.LSPNQVVIV |
| 6.LAESRSMFF | 29.VCQHIDMMQ | 52.WRAMDMLCP | 75.VVIVSCLVH |
| 7.MFFLKHTVD | 30.IDMMQTHDM | 53.MLCPEMEHA | 76.VIVSCLVHV |
| 8.FFLKHTVDT | 31.MMQTHDMKA | 54.LCPEMEHAP | 77.IVSCLVHVG |
| 9.FLKHTVDTF | 32.LSDMETKFT | 55.MEHAPALFI | 78.VSCLVHVGG |
| 10.LKHTVDTFR | 33.METKFTIWE | 56.FIDWAAGTA | 79.VGGFGHINW |
| 11.VDTFRKLFV | 34.FHQPALADA | 57.IDWAAGTAI | 80.INWEKMHGI |
| 12.LFVLMVFHQ | 35.LADANSESS | 58.WAAGTAIKW | 81.MHTEVEHIV |
| 13.FVLMVFHQG | 36.VHLECRQKQ | 59.IKWCPDEWC | 82.VEHIVKEDT |
| 14.VFHQGNGLV | 37.LECRQKQPR | 60.MCREQNCVC | 83.IVKEDTEKG |
| 15.FHQGNGLVH | 38.CRQKQPRGC | 61.VCSQNTMST | 84.VKEDTEKGS |
| 16.LVHAMCGVW | 39.PRGCMVVHV | 62.VFTTRHRVM | 85.WCWLRAIQC |
| 17.VHAMCGVWH | 40.VVHVQRIEN | 63.FTTRHRVMS | |
| 18.MCGVWHWCL | 41.QRIENHIPH | 64.WALEFNEMM | |
| 19.VWHWCLDHD | 42.IENHIPHQC | 65.LEFNEMMAK | |
| 20.MALCIRMCG | 43.WIKGIPQMW | 66.MMAKFDHKT | |
| 21.LCIRMCGKG | 44.MWEASHLGH | 67.FDHKTRPDT | |
| 22.IRMCGKGCQ | 45.WEASHLGHM | 68.TRPDTANAR | |
| 23.MCGKGCQKN | 46.IFNWQWHDK | 69.ARCVGMVVM | |
El espacio muestral obtenido contiene 209 posibles microestados, péptidos nonaméricos, asociados a los 20 aminoácidos esenciales; de estos existen 20 microestados que cumplen la característica de tener el mismo aminoácido en todas sus posiciones, por lo tanto su probabilidad es 20/209=3.9x10-11; mientras que existen 206 microestados que cumplen la característica de tener, en las tres primeras posiciones, un aminoácido definido -por ejemplo triptófanos-, y en las posiciones restantes cualquier aminoácido esencial, lo cual lleva a una probabilidad de 206/209=1.25x10-4. El cociente entre las probabilidades calculadas es 3,12x10-7, lo cual muestra una diferencia entre el péptido presentado y el péptido teórico de no unión; en órdenes de magnitud hay 10 millones de microestados con las tres primeras posiciones fijas por cada microestado con todos los aminoácidos iguales.
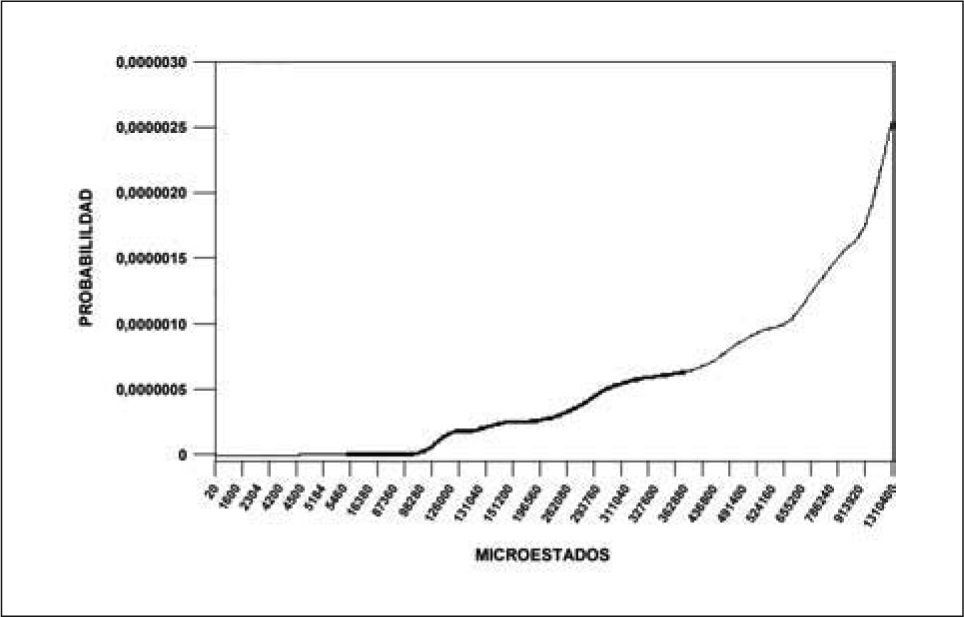
El cálculo de las probabilidades y el número de microestados asociados a los tipos de secuencia seleccionados para el paso 1 y su posterior evaluación con los criterios definidos (paso 2), se muestran en la Tabla V y la Figura 1. Los cálculos de probabilidad, número de microestados, para el paso 3 y su posterior evaluación con los criterios definidos en el paso 4, se muestran en la Tabla VI. Los valores de probabilidad para todos los tipos de secuencia en el paso 1-2, oscilaron entre 3,9x10-11 y 1,1x10-3, la probabilidad calculada para el CLIP y el HA es de 1,01x10-8 y 7,08 x10-7, respectivamente; la región limitada por estos valores de probabilidad está dentro de un rango característico de unión. Los valores de probabilidad en el paso 3-4, oscilaron entre 2,10 x10-9 y 7,03 x10-8.

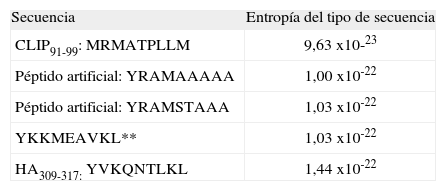
El cálculo del valor de entropía para cada tipo de secuencia aplicando el paso 1 y su posterior evaluación con los criterios definidos (paso 2) se muestran en la Tabla VII y la Figura 2. Los valores de entropía para el paso 3 y su posterior evaluación con los criterios definidos (paso 4), se muestran en la Tabla VIII.

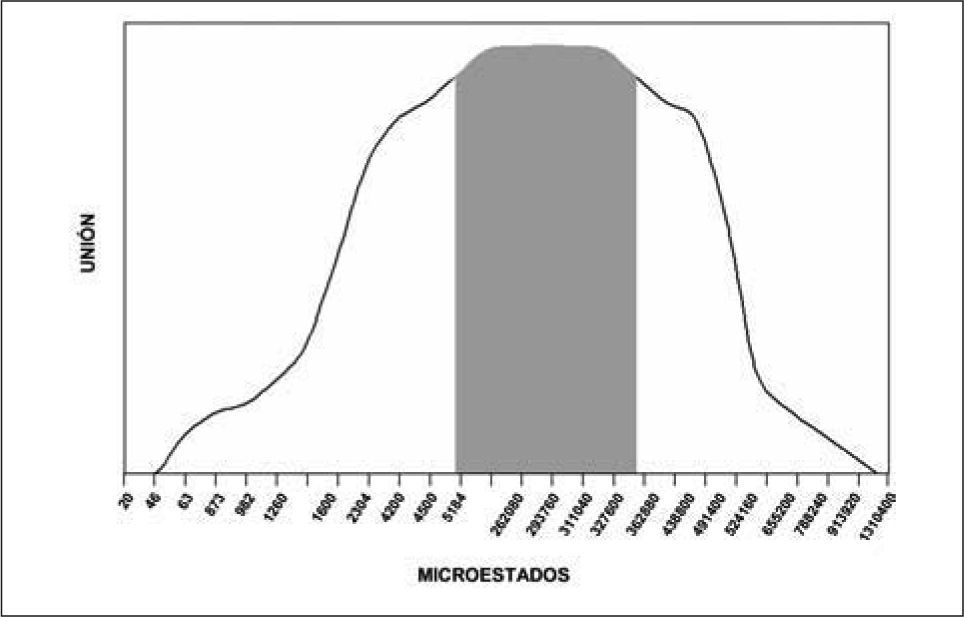
Se encontró que en el paso 1-2 todos los posibles péptidos de unión están incluidos en los tipos de secuencia que presentan valores de entropía comprendida en el rango [1,18x10-22, 1,76x10-22], mientras que los péptidos de no unión están incluidos en los tipos de secuencia que presentan valores de entropía mayores que 1,76x10-22 o menores que 1,18 x10-22. En el paso 3-4 los tipos de secuencia asociados a la unión tienen valores de entropía comprendida en el rango [9,63x10-23, 1,44x10-22].
A partir de los cálculos realizados se observa que el valor mínimo de entropía corresponde a las secuencias que tienen un único aminoácido, mientras que la máxima entropía corresponde a secuencias con todos los aminoácidos diferentes; siendo estos extremos los casos en que no se presenta unión por parte del péptido, mientras que los tipos de secuencia asociados a unión tienen valores intermedios de entropía.
Para el tipo de secuencia asociado al CLIP, se encontró que posee 5184 microestados equivalentes y que la proporción S/k es 1,18x10-22 / 1,38x10-23=8,55, mientras que para el tipo de secuencia asociado al HA que posee 326880 microestados equivalentes, la proporción S/k es 1,76x10-22 / 1,38x10-23=12,8 (Tablas IX y X).
La metodología desarrollada en este trabajo permite predecir cadenas nonámeras de péptidos que se unen al HLA clase II en la región central para cualquier péptido sintetizado o natural, a partir de las características matemáticas establecidas para la diferenciación de péptidos que se unen de los que no. De los 161 péptidos probados, la predicción de macroestados de unión y no unión acertó en un 100%. A partir de la relación S/k se deduce matemáticamente el porcentaje de péptidos asociados a la unión con respecto a la totalidad de los tipos de secuencia posibles, encontrando valores de 15.8% para el paso 1-2 y 13% para el paso 3-4 (Tablas IX y X). Para la secuencia teórica se encontraron 492 péptidos nonaméricos sobrelapados, al calcular sus valores de probabilidad, combinatoria, y entropía se encontraron 85 regiones centrales asociadas al macroestado de unión y 407 al de no unión (Tabla XIII, Anexo 3).
DISCUSIÓNEste es el primer trabajo donde se realiza una teoría de predicción de unión de péptidos presentados por moléculas de HLA clase II basada en leyes de probabilidad, de combinatoria y de entropía. Se encontraron valores numéricos de probabilidad y de combinatoria que caracterizan matemáticamente cualquier posible microestado. Con la relación S/k y la ecuación más simple de entropía se predijeron cuales son los valores que corresponden al macroestado de unión.
Los valores de probabilidad y de combinatoria permitieron diferenciar y caracterizar matemáticamente los péptidos que se unen de los que no. Los valores de entropía calculados se encuentran dentro de valores finitos para cualquier cadena nonamérica, similar a la generalización de la entropía desarrollada por Bekenstein(25), donde no hay valores infinitos para la entropía. Además, estos valores no definen una flecha unidireccional en el fluir del tiempo, pues no evalúan la evolución del sistema. A partir de la ley de entropía se obtiene la relación S/k, la cual posee proporciones de una ley física de la naturaleza que depende de la forma en que los microestados W, tienen un orden de combinación, permitiendo predecir físicamente los péptidos nonámeros de unión y no unión. Las predicciones de entropía comprueban cómo péptidos construidos sintéticamente son presentados por la molécula de HLA clase II, evidenciando que esta teoría puede generar teóricamente y predecir cualquier posible experimento diseñado para unión. La relación S/k como el valor predicho, en un rango finito, comparado con el rango total de valores posibles, demuestra matemáticamente la alta especificidad del fenómeno de unión en la naturaleza comparada con todas las posibilidades matemáticas. Desde esta nueva perspectiva teórica comparada con el trabajo experimental de la inmunología y basados en las predicciones de los péptidos posibles de unión de la proteína teórica desarrollados, es muy posible predecir péptidos que se unan a la molécula de HLA clase II que jamás hayan sido probados experimentalmente, similarmente a la mecánica Newtoniana a partir de la cual se predijo la existencia de un planeta(26) que no había sido visto previamente, lo que elevaría la inmunología a nivel de ciencia fundamental en el caso de la presentación por las moléculas de HLA clase II.
Se calculó el número de microestados para cada tipo de secuencia y la entropía asociada a este número, partiendo de observaciones experimentales que permitieron desarrollar generalizaciones. Desde esta perspectiva teórica se definieron 10 criterios evaluadores para el paso 2 y 10 criterios para el paso 4 que permitieron caracterizar la especificidad del macroestado de unión. Los cinco primeros para el paso 2 y los dos primeros del paso 4 definieron la especificidad para las 24 regiones centrales estudiadas(23) y los siete macroestados seleccionados, mientras que los demás criterios permitieron refinar los cálculos, esto enmarcado desde las leyes teóricas aplicadas para calcular microestados particulares.
En las Tablas VI y VII se observa cómo la ley de entropía, que depende de W y es creciente, caracterizó los aminoácidos que se unen al HLA de clase II y los diferenció de los que no; lo cual se corrobora. Por ejemplo, al CLIP se asocian 1080 microestados y tiene una entropía de 1,18 x10-22, lo cual corresponde a la predicción de unión, mientras que el péptido GSVQYPITK con 604'800.000 microestados tiene una entropía de 2,79 x10-22 y corresponde a la predicción de no unión. Al deducir la ecuación 3 mediante un proceso algebraico, que conserva la relación funcional de sus variables, es posible predecir el número de microestados que tendría un péptido que se una al HLA clase II. De la misma manera, la ecuación de la entropía en este contexto estudia la manera en la que conjuntos de aminoácidos se asocian en un orden, análogo a un sistema de partículas en el estudio de la materia, y el resultado general caracteriza la unión o la no unión de esos conjuntos de aminoácidos. En conjunto, la teoría desarrollada lleva a la predicción de la existencia o inexistencia de secuencias de aminoácidos, como en la secuencia teórica de un mismo aminoácido en las nueve posiciones, y la diferenciación con secuencias de aminoácidos diferentes evaluando de aquellos que se pegan de los que no.
Trabajos anteriores en predicción de unión de péptidos han utilizado Artificial Neural Networks, ANN, programación lineal, o vectores de máquina(27-30); estos métodos buscan patrones de unión caracterizando las posiciones y sus respectivos motivos. Su generalización es limitada, pues depende de la muestra usada. La teoría desarrollada en este trabajo confirmó los resultados experimentales, en términos de su representación numérica y de las combinaciones de aminoácidos apropiadas. A diferencia de los trabajos mencionados, el presente es de naturaleza teórica e independiente de la población de estudio. Ahora bien, el método experimental de péptidos sobrelapados, al que Berzofsky definió como "método de fuerza bruta"(31), consiste en la síntesis de péptidos, pertenecientes a la secuencia de una misma proteína que se sobrelapan. La capacidad de dichos péptidos para estimular respuestas en los linfocitos T, tanto proliferativas como citotóxicas, es probada luego in vitro. Aunque este método es completo, es costoso e implica mucho tiempo y trabajo. La teoría planteada en este trabajo permite evaluar todas las secuencias nonámeras sobrelapadas para cualquier secuencia proteica, simplificando el método empleado pues se dejan a un lado los procesos experimentales de ensayo y error por predicciones numéricas rigurosas.
Trabajos experimentales evidenciaron pérdida de la energía de unión debido a la disminución del tamaño del péptido asociado a la molécula de HLA clase II, el cual generalmente varía entre 13 y 16 aminoácidos(15-18). Una compensación a ésta energía perdida es evidenciada a través de las interacciones de la región central, y puede ser provista por la adición de aminoácidos en las posiciones de anclaje(23). Desde estos resultados experimentales sería posible diseñar antígenos sintéticos que se presenten a las moléculas de HLA clase II con las longitudes predichas por esta teoría, o determinar el lugar de la región central de la secuencia antigénica sin importar su longitud. Desde este punto de vista propongo predecir los posibles péptidos nonaméricos de unión naturales o sintéticos; hacer la contrastación experimental de unión; y posiblemente introducir aminoácidos que cambien los valores de los microestados, buscando el diseño de péptidos promiscuos, lo importante del tercer y cuarto paso, de acuerdo a las evaluaciones matemáticas. Finalmente, hacer las pruebas experimentales de respuesta y memoria inmunológica y así poder desarrollar vacunas a partir de esta metodología física y matemática.
Este trabajo muestra el desarrollo de una teoría a partir de tres leyes aplicadas, evidenciando órdenes físicos y matemáticos subyacentes a la presentación antigénica. La teoría desarrollada logra simplificar este fenómeno tan complejo, partiendo de solo siete péptidos con los que se construye la generalización que obvia la especificidad alélica; pues el fenómeno de unión es caracterizado a partir de representaciones numéricas de tipos de secuencias y no de aminoácidos específicos. Como en las teorías fundamentales actuales de la física(32) no hay causas sino órdenes físicos y matemáticos acausales para la descripción y comprensión del fenómeno estudiado. Por ser una teoría no requiere de análisis estadísticos ni del uso de grandes cantidades de datos experimentales.
FUTURAS APLICACIONESSe contrastará esta teoría con las bases de datos experimentales desarrolladas y se comparará con los modelos predictivos actuales. Se compararán predicciones de secuencias antigénicas u oncogénicas de proteínas con péptidos que se conoce son presentados por moléculas de HLA clase II para la corroboración de esta teoría.
CONCLUSIONES- 1.
Las probabilidades y las combinatorias para todos los microestados posibles se encuentran dentro de un rango finito caracterizando de forma objetiva y reproducible la unión.
- 2.
Los valores de entropía para todos los microestados posibles se encuentran dentro de un rango finito y predicen todos los valores asociados a el macroestado de unión y al de no unión. La evidencia matemática de la alta especificidad del fenómeno de unión en la naturaleza está representada por el valor predicho que debe tener el sistema para que se una, el cual está dado por la relación S/k.
- 3.
Este estudio permitió encontrar órdenes matemáticos subyacentes al fenómeno de unión del HLA clase II superando los procesos experimentales de ensayo y error. La predicción física y matemática realizada en la presente investigación facilita el proceso de selección de los péptidos requeridos en el desarrollo de vacunas; optimizando el tiempo y recursos económicos.
- 4.
La predicción teórica de secuencias de péptidos naturales no evidenciados experimentalmente puede ser posible.
El autor declara no tener ningún conflicto de interés financiero.
A todos los que me ayudaron y a todos los que me rechazaron, por que sin todos no hubiera sido posible llegar. Infinitas gracias a todos y a todo. Infinitas gracias a todos los que están y estuvieron en el Grupo Insight.
Dedicado a Sir. Isaac Newton, MAESTRO del camino.