


La inflamación es un proceso fisiológico dirigido a prevenir o reparar los daños producidos por agresiones externas, infecciones o intoxicación. Este proceso depende de la coordinación entre la activación y migración de los leucocitos hacia la región dañada y por tanto está asociado con cambios en la morfología celular y con la reorganización del citoesqueleto de actina. El citoesqueleto de actina también contribuye a otros cambios morfológicos esenciales necesarios para la vida de la célula. WIP (WASP Interacting Protein) es una proteína que se une a la actina y participa en procesos inflamatorios. Dicha unión estabiliza los microfilamentos de actina y regula su organización especial y temporal. La ausencia de WIP induce la formación de redes de actina deficientes en linfocitos T y B, lo que conduce a la modificación de la capacidad linfocitaria de activación y migración y desemboca en alteraciones autoinmunes con infiltración leucocitaria en órganos vitales y muerte del animal.
Inflammation refers to a physiological process aimed at preventing or repairing the damage induced by infection, injury or intoxication. This process requires the coordinated activation and migration of leukocytes to the damaged area and therefore it is associated with changes in cell morphology and actin cytoskeleton reorganization. Actin cytoskeleton is also required for many other changes in cellular morphology that take place during cell life. WIP (WASP Interacting Protein) is an actin-binding protein involved in inflammation. WIP stabilizes actin filaments and regulates their temporal and spatial organization. Absence of WIP leads to formation of defective actin networks in T and B lymphocytes (that entail defective lymphocyte activation and migration) and ultimately results in autoimmunity and leukocyte infiltration in vital organs resulting in a fatal outcome.
The three principal roles of a functional immune system could be summarized as: detection of foreign antigens not previously encountered, amplification of responses to previously detected antigens and delivery of coordinated effector responses ranging from mobilization of effector cells, to cytokine delivery and antibody production(1). Accordingly, the efficiency of the immune response depends upon organized cellular homing and migration within lymphoid organs, antigen recognition and cell signaling and activation. It is also necessary to avoid disorganized and delocalized responses that could induce autoimmune attacks against self-antigens. A common event underlying each of these processes is the reorganization of the actin cytoskeleton. Gene deletion approaches as well as pharmacological studies have shown that actin reorganization is required for the initial steps of lymphocyte activation (receptor translocation to the cell surface to facilitate conjugate formation, initiation and continuation of signaling), as well as for later steps involving cell shape modification (polarization and cell translocation). Consequently, in recent years there has been an increasing interest in understanding the molecular basis of the process of actin polymerization, as well as in the contribution of the proteins involved in its regulation in order to regulate the immune response. This review will focus on the role in immune cell activation and migration of some of the best-studied actin regulatory proteins in immune cells: WASP (Wiskott Aldrich Syndrome Protein) and its binding partner WIP (WASP Interacting Protein).
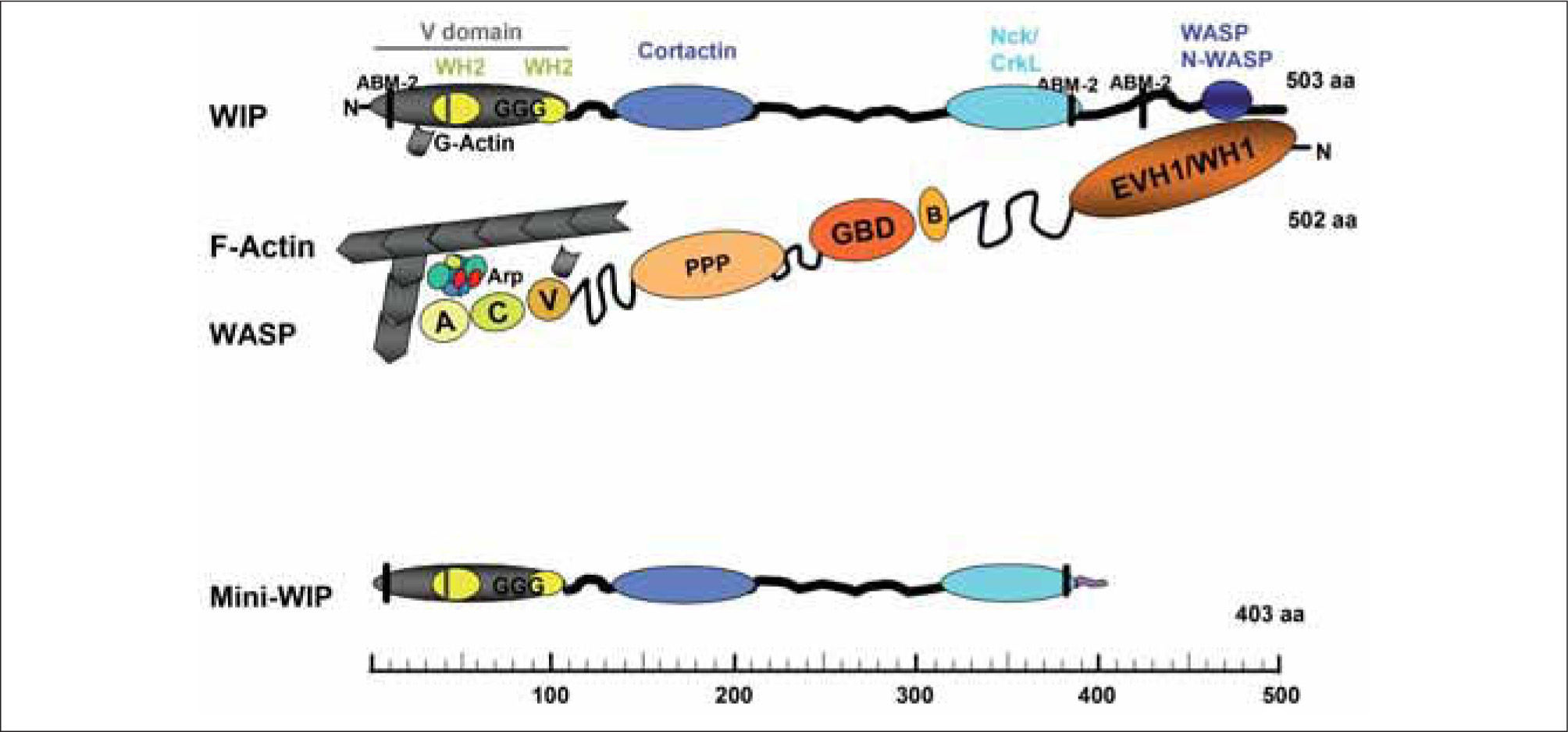
WIP FAMILY OF PROTEINSThe gene coding for WIP is located on chromosome 2 in humans and mice and codes for a 503 amino acid (aa) protein or a 493 aa protein, respectively. WIP is an actin-binding protein widely conserved along evolution: the expression of WIP or its orthologs has been described in mold, yeast, invertebrates and vertebrates(2). The mammalian WIP family of proteins includes three members: CR16 (glucocortocoid-regulated protein)(3), WIP(4), and WICH/WIRE (WIP-CR16 homologous/WIP-related)(5,6). All three members share around a 30% sequence identity, an unusually high percentage of proline residues (betwen 27% and 32%) and two highly homologous regions (Fig. 1): the verprolin homology region (V-domain) located near the N terminus and the WASP/N-WASP (neural WASP) binding domain (WBD) located at the C-terminus. The V-domain includes two actin-binding WH2 (WASP homology 2) domains, and in WIP there is also a polyglycine-rich sequence. CR16, WIP and WHICH/WIRE bind both monomeric globular actin (G-actin) and filamentous actin (F-actin), and contain between three and six actin-based motility homology-2 (ABM-2) motifs, potential binding sites for profilin (an actin polymerization regulating protein). WIP binding to cortactin, to the adapters Nck-1 and CrkL(7,8)(Fig. 1), and to the Src family tyrosine kinase Hck (hematopoietic cell kinase)(9) has been also demonstrated. The tissue distribution of WIP differs significantly from that of CR16 and WICH/WIRE. CR16 is found predominantly in the brain and at lower levels of expression in the lung, heart(10) and testes(11) and WICH/WIRE expression is mainly detected in colon, lung and stomach(6). In contrast, WIP is widely expressed and, at the protein level, it has been identified at low amounts in fibroblasts and endothelial cells and at significantly higher levels in haematopoietic tissues, including lymphoid organs and immune primary cells or cell lines (lymphocytes, mast cells, natural killer (NK), dendritic cells (DC) and osteoclasts)(2). Interestingly, a recently described truncated form of WIP (termed mini-WIP) lacking the WASP/N-WASP binding site is expressed in peripheral blood cells but not in fibroblasts(12). This shorter form could act as a regulator of full length WIP function by competing its binding to certain partners(12).
, including one of the three actin-based motility 2 domains (ABM-2) for profilin binding, two WASP homology 2 (WH2) domains for actin binding, and a polyglycine-rich sequence (GGG). The V domain is followed by a multitude of proline-rich motifs that bind cortactin, Nck or CrkL and at the C-terminus resides the WASP/N-WASP binding site (WASP, N-WASP). Mini-WIP, a WIP isoform, lacks the WASP-binding site and its C-terminal sequence differs from that of WIP. Actin and Arp2/3 complex (Arp, actin related protein) bind to the VCA region. WIP is bound to the EVH1/WH1 domain of WASP so their actin-binding regions cooperate for microfilament polymerization. Note that WIP and WASP are represented in opposite sense, that is, WIP N-terminus is on the left and WASP N-terminus is on the right. Abbreviations: WASP: EVH1, Ena/Vasp homology 1 domain; WH1, WASP homology domain 1; B, basic domain; GBD, GTPase binding domain; PPP, proline rich domains; VCA, verpolin cofilin homology domains/acidic region.")
WIP and WASP form a heterodimeric complex involved in the regulation of actin polymerization. Domain organization of WIP, showing the major protein interaction sites. Verprolin homology domain (V domain), including one of the three actin-based motility 2 domains (ABM-2) for profilin binding, two WASP homology 2 (WH2) domains for actin binding, and a polyglycine-rich sequence (GGG). The V domain is followed by a multitude of proline-rich motifs that bind cortactin, Nck or CrkL and at the C-terminus resides the WASP/N-WASP binding site (WASP, N-WASP). Mini-WIP, a WIP isoform, lacks the WASP-binding site and its C-terminal sequence differs from that of WIP. Actin and Arp2/3 complex (Arp, actin related protein) bind to the VCA region. WIP is bound to the EVH1/WH1 domain of WASP so their actin-binding regions cooperate for microfilament polymerization. Note that WIP and WASP are represented in opposite sense, that is, WIP N-terminus is on the left and WASP N-terminus is on the right.
Abbreviations: WASP: EVH1, Ena/Vasp homology 1 domain; WH1, WASP homology domain 1; B, basic domain; GBD, GTPase binding domain; PPP, proline rich domains; VCA, verpolin cofilin homology domains/acidic region.
In the last ten years WIP involvement in a myriad of activities in diverse cell types has become the focus of great interest(2). In this article we will concentrate on WIP activities that are directly linked to its role in actin polymerization in immune cells and therefore could contribute to define the role of WIP in leukocyte activation and migration.
WIP contributes to the stabilization of actin filaments and enhances actin bundling(13). These capabilities could explain the increase in polymerized actin observed in transfected human B lymphocytes over-expressing WIP(4). In Jurkat cells increased WIP levels also results in augmented WASP, but not N-WASP(14). Conversely, a reduced WIP expression (as described either in lymphocytes and DC derived from WIP-deficient mice, or in cell lines expressing interference RNAs to silence WIP expression) produces severely decreased levels of WASP(14–16). The WASP- binding domain of WIP protects WASP from degradation by the calcium activated protease calpain in vitro. The smallest WIP fragment described to efficiently prevent WASP degradation is comprised of aa 410-503. Structural data support that WIP wraps around folded WASP contacting at several defined aa sequences(17). Thus, under physiological conditions the vast majority of WIP and WASP molecules exist as obligate cytoplasmic heterodimers (i.e. in T lymphocytes 90% of expressed WIP is bound to 75-95% of WASP)(8,14). Not surprisingly, most missense mutations in WASP that have been shown to be responsible for causing the severe immunedeficiency WAS and the milder form X-linked thrombocytopenia, are located in the WIP binding WH1 (WASP homology 1) domain, and some of them have been demonstrated to impair WIP binding and to diminish WASP stability(14,18).
WIP contributes to maintain the autoinhibited form of WASP mediated by intramolecular interaction between the Cterminal VCA region and the GBD (GTPase binding domain) domain and its surrounding regions(19) (Fig. 1). This complex translocates to cellular areas where actin remodeling will occur following receptor engagement and signal transduction as it has been demonstrated in T lymphocyte, NK cells and DCs(8,15,20). The importance in the immune system of the formation and proper localization of a WIP-WASP complex has been extensively described and will be discussed below. In the human monocyte cell line THP-1, immunoprecipitation with anti-WASP brings not only WIP but also WICH/WIRE(21), pointing to the existence of a multiprotein complex involved in actin cytoskeleton reorganization. Cellular activation (e.g. through TCR in T lymphocytes) induces Nck recruitment to the plasma membrane, PIP2 (phophatidylinositol (4,5)-biphosphate) production, and induction of the active form of the RhoGTPase Cdc42 that contributes to the release of the intramolecular interaction of WASP. The open form of WASP activates the nucleation activity of Arp 2/3 complex and promotes actin polymerization and reorganization. Hence, it would be expected that the absence of WIP would facilitate WASP activation and ultimately actin polymerization. In contrast, WIP deficiency leads to severe defects in several cellular functions mediated by actinpolymerization following WASP activation, such as T-cell signaling, mast cell degranulation, lymphocyte homing and chemotaxis and podosome formation(2). Similar results are obtained when WIP-deficient cells, either derived from WIP−/− mice or from wild type primary cells or cell lines that express interference ARN to diminish WIP levels, are analyzed. These results exclude the possibility of an artifact due to manipulation of stem cells or homologue recombination required for mouse generation, and provide evidence to support that WIP function goes beyond inhibition of WASP/N-WASP activation.
The functional WIP/WASP complex is not restricted to populations of immune system cells in mammalian organisms. Data derived from studies of the formation of syncytial muscle fibers in the Drosophila system supports the existence of a complex including the fusion-competent cell-specific receptor Sns, solitary/D-WIP and Wsp (Drosophila orthologs of WIP and WASP, respectively)(22,23).
In contrast to the above described WIP function as a promoter of WASP stability and therefore as a promoter of actin polymerization, WIP also has a role as a negative regulator of WASP/N-WASP. WIP maintains N-WASP/WASP in an inactive state in resting cells(24), and inhibits Cdc42-mediated N-WASP-induced activation of the actin nucleating Arp2/3 complex in vitro(25). This inhibition does not persist in vitro in the presence of PIP2, another activator of WASP/N-WASP that contributes to the release of the intramolecular interactions and favors Arp2/3 activation. The recently described contribution of another player may explain these apparently contradictory functions of WIP. In cells, N-WASP activation also requires the contribution from TOCA-1 (transducer of Cdc42 mediated actin assembly-1), a protein that binds both Cdc42 and N-WASP and relieves WIP-mediated inhibition, allowing Cdc42 activation of the N-WASP/WIP complex(24).
In summary, WIP plays important roles in actin polymerization, both dependent and independent on its binding to WASP. WIP mediates WASP effect as a microfilament regulator by: 1) protecting WASP from degradation by proteases, 2) regulating WASP and N-WASP activation by Cdc42, TOCA-1 and PIP2, and 3) contributing to WASP translocation to areas of active actin reorganization.
LYMPHOCYTE ACTIVATIONReorganization of the actin cytoskeleton is an essential component in T lymphocyte activation following the interaction between the T cell receptor (TCR) and peptide-loaded major histocompatibility complex (pMHC) molecules on the surface of antigen-presenting cells (APC)(26,27) as well as on NK cells(20). Receptor engagement causes an increase in the cellular F-actin content as well as a distinctive pattern of F-actin distribution that contributes to characteristic changes in cell shape(28). The recognition of antigenic peptide by T cells entails the coordinated movement of TCR and other molecules, including the integrin lymphocyte function-associated antigen-1 (LFA-1) that binds to intercellular adhesion molecule-1 (ICAM-1)(29). The specialized cell surface junction formed in a T lymphocyte interacting with APC is named "immunological synapse" (IS)(30) and is composed of a central supramolecular activation cluster enriched with TCRs surrounded by a ring of adhesion molecules (peripheral supramolecular activation cluster). The formation of the IS is an actin-dependent process that causes TCR and adhesion proteins to cluster at the cell periphery after an initial segregation from one another at the earliest stages of microdomain formation. The TCR and adhesion microdomains attach to actin and are carried centripetally by retrograde flow. The TCR microdomains penetrate into the actin-depleted cell center, whereas the adhesion microdomains appear to be unstable without an underlying actin cytoskeleton. Finally, TCR and adhesion molecules spatially partition from one another well before the formation of a mature IS by differential actin interactions that help to shape and maintain the final bull's-eye pattern of the IS(31). Altered spatial organization of synaptic proteins directly affects downstream signaling, implying that spatial reorganization of the TCR and associated signaling molecules serves as an active regulatory mechanism(19–21). Very recently, the dynamic portrait of repatterned immunological synapse formation has allowed to propose a mechanism for spatially sorting T cell surface molecules that only relies on their relative coupling strength to actin flow. This mechanism is based on a frictional or viscous coupling to actin flow that allows slipping of diverse molecules(32).
WIP contributes to IS formation in T lymphocytes and NK cells(20,33). Electron microscopy analysis of the subcortical actin network of lymphocytes purified from WIP-deficient mice reveals that WIP is critical for the integrity of the actin cytoskeleton in both T and B lymphocytes(33). However, while WIP deficiency impedes T lymphocyte activation, it promotes B lymphocyte activation, pointing to an opposite role for actin cytoskeleton and WIP in T versus B lymphocyte activation. T lymphocytes from WIP-/- mice do not respond to activation with plate-bound anti-CD3 plus co-stimulatory anti-CD28: WIP-/- T lymphocytes neither proliferate nor secrete the autocrine growth factor interleukin (IL)-2. Interestingly, absence of WIP does not significantly affect early signaling that follows TCR engagement as assayed by protein tyrosine phosphorylation or calcium flux. In contrast, absence of WIP leads to the formation of smaller contact areas between T cells and structures bearing TCR activating molecules, being those anti-CD3 antibody in planar bilayers and superantigen-loaded APC in assays of IS formation. Depletion of WIP by RNA interference in the NK cell line YTS prevents the formation of a multiprotein complex (consisting of WIP, WASP, actin and myosin IIA in normal conditions) and leads to almost complete inhibition of cytotoxic activity(20) due to impaired lytic granule polarization(34). All these data support a critical role for WIP in T, B and NK cell IS formation and activation.
A reasonable simplifying assumption to explain the T cell pathology in the absence of WIP was that the defect in IL-2 secretion was secondary to defects in actin polymerization. However, a growing body of evidence indicates that actin polymerization and promotion of IL-2 transcription are largely distinct functions of the WASP/WIP complex. The data supporting this statement are the following: 1) WIP can synergize with the guanine nucleotide exchange factor Vav to enhance activation of NF-AT/AP-1 transcription elements within the IL-2 promoter through both WASP-dependent and WASP-independent pathways(35); 2) Deletion of the verprolin/cofilin/acidic domain COOH terminus of WASP hinders its function in actin polymerization but facilitates its role in IL-2 transcription in T cells as assessed by NF-AT reporter assays(36); 3) The NH2 terminus of WASP (including the WH1 domain) functions as dominant negative for proliferative responses and cytokine production induced by TCR stimulation, but not for actin polymerization(37), similarly to what it is observed in transgenic mice expressing anti-WH1 scFvs (single chain fragment variable) intrabodies(38); 4) Analysis of T cells from WASP-deficient mice shows a major effect on IL-2 production but only subtle alterations in synapse formation(39). Moreover, in NK cells the WASP- regulated nuclear translocation of NF-AT2 andNF-κB (RelA) occurs independently of its role in filamentous actin polymerization and actin cytoskeletal rearrangement(40,41); 5) Dong and coworkers have identified that the highly conserved NH2 terminus of WIP contains a region that is strongly inhibitory for NF-AT activation, with contributions from both an NH2-terminal polyproline region and the actin binding NH2-terminal WH2 domain, and suggests that WIP, like WASP, is subject to autoinhibition(42). In summary, the data suggest that WIP stabilization of WASP explains the otherwise unexpected results in TCR-induced NF-AT activation.
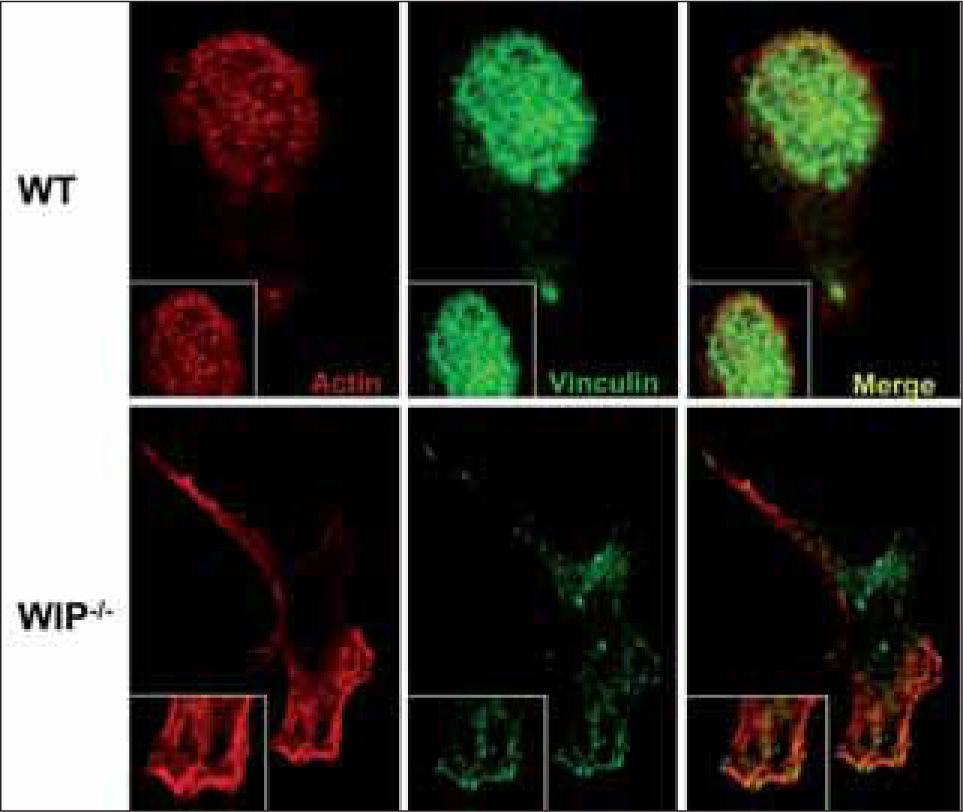
MIGRATIONAn efficient immune response relays on regulated cell migration in terms of temporal and spatial distribution. Cell migration is not only required during the development of the immune system (i.e., when cells must translocate between immune organs for differentiation and maturation), but also for immune effector functions. For example, activated endothelial cells and tissue fibroblasts produce and secrete chemoattractants at the site of injury aimed at recruiting effector phagocytic leukocytes, macrophages and neutrophils that consequently migrate to the injured or infected area. Impaired migration can cause defects in monocyte and leukocyte recruitment to inflammed tissues, resulting in recurrent infections. In WAS patients, recurrent infections are suggested to be due to chemotactic defects reported in hematopoietic cells such as neutrophils, monocytes/ macrophages and dendritic cells(43). Very recently, a role for WIP and WICH/WIRE in the chemotaxis of monocytes has been described. When binding of WASP to these proteins is blocked, actin polymerization takes place but cell polarization is impaired(43). As a result of altered WASP localization due to the absence of WIP or to impaired binding to WIP, actin polymerization occurs at inappropriate sites resulting in dysfunctional actin-rich structures. Similarly, overexpression of WASP in DCs lacking WIP promotes the formation of actin clusters instead of the functional podosomes observed in normal DC (Fig. 2)(15). These data suggest that the WASP/WIP complex acts as a unit to establish cell polarization required for monocyte chemotaxis.
 and WIP−/− DCs plated on poly-L-lysine coated glass coverslips were doublestained to identify the formation of podosomes and the distribution of vinculin and F-actin by confocal microscopy. Podosomes are grouped in clusters in WT DCs while large focal contacts containing vinculin comprise the most striking adhesion structures in WIP+ DCs, replaáng podosomes as the major points of contact with the substratum.")
WIP−/− DCs are devoid of podosomes. Wild type (WT) and WIP−/− DCs plated on poly-L-lysine coated glass coverslips were doublestained to identify the formation of podosomes and the distribution of vinculin and F-actin by confocal microscopy. Podosomes are grouped in clusters in WT DCs while large focal contacts containing vinculin comprise the most striking adhesion structures in WIP+ DCs, replaáng podosomes as the major points of contact with the substratum.
Another essential mechanism required to complete a coordinated and efficient immune response is lymphocyte homing. Purified splenic T lymphocytes from WIP−/− mice show deficient in vivo homing to spleen and lymph nodes one hour after intravenous inoculation into wild type recipients(44). This chemotactic defect is confirmed in transwell assays in response to SDF-1a (stromal cell-derived factor-1 a), a chemokine involved in the migration of T lymphocytes across lymph node high endothelial cells and lymphoid organs in vivo(45,46). This defect is not due to decreased rolling, integrin-mediated adhesion, or CXCR4 expression, but to abnormal reorganization/redistribution of the newly polymerized F-actin in response to SDF-1α stimulation. More specifically, there is a significant increase in the number of bipolar cells in WIP−/− T cells following stimulation. Interestingly, mice deficient for both WASP and WIP show significantly more severe defects in T-cell homing and chemotaxis than either of the single knockouts, pointing, on one hand, to a degree of functional redundancy between the two proteins, and on the other hand to a role for WIP in T cells independent on its effects on WASP. Disruption of WIP/WASP complexes by means of overexpression of a WIP fragment including the WASP binding domain, also results in impaired transendothelial migration of human primary macrophages through HUVEC (human umbilical vascular endothelial cell) monolayer(21).
Cell migration comprises a cycle of several highly coordinated and regulated steps(47). Directional movement is initiated by polarization of the cell and membrane protrusion at the front to form the leading edge. Subsequently, there is a second step aimed at forming adhesion complexes that link the extracellular matrix to the actin cytoskeleton, allowing the generation of the forces necessary to translocate the cell body forward. Eventually, adhesive contacts at the rear end disassemble to allow cell detachment and net cell translocation. This repeated migratory cycle relays on consecutive rounds of formation and disassembly of adhesion complexes, some of them regulated by calpains. The calpain family of proteases contribute to the control of cell adhesion and migration through their ability to degrade key components of adhesion structures and of the actin polymerizing machinery, including β integrins, paxillin, vinculin, cortactin and WASP(48).
PROTEIN DEGRADATION: CALPAIN AND/OR PROTEASOME. PODOSOME FORMATIONThe calpain family of proteins are calcium-dependent cysteine proteases that cleave substrates between protein domains in relatively unstructured regions of proteins lacking primary aa consensus sequences(48). This activity can be specifically inhibited by the ubiquitously present calpain inhibitor calpastatin. In a muscle system, calpain activity has been shown to increase proteasome-dependent proteolysis, another protein degradation route in cells(49). This large ATP-dependent proteolytic complex is the main site for protein degradation in mammalian cells and catalyses the rapid degradation of ubiquitinated proteins. The 26S component of the proteasome complex recognizes target proteins containing the 76 aa peptide ubiquitin, covalently linked to lysine residues. Since ubiquitin itself contains seven lysine residues, ubiquitin molecules can generate different types of polyubiquitin chains. Ubiquitinylation of N-WASP has been described in primary cortical neurons and neuronal cell lines(50). This posttranslational tag marks N-WASP for degradation by the proteasome. Interestingly, the absence of WIP increases WASP susceptibility to degradation by both, calpain and proteasome complex. Initial observations pointing to the role of WIP as a chaperone for WASP integrity were derived from the analysis of WASP levels in cells obtained from WIP−/− mice(44). Absence of WIP led to a severe reduction of WASP in T and B lymphocytes (retaining around 10% of control WASP), and dendritic cells(14,15,44). Confirmation of this role for WIP came from the observation that silencing of WIP expression using lentiviral vectors producing shRNA specific for WIP mRNA in Jurkat cells resulted in decreased WASP levels, without modifying N-WASP levels. Interestingly, neither WIP nor WASP are detectable in the erythroleukemic line K562 and significant expression of exogenous WASP by retroviral-mediated gene transfer was only achieved by cotransfection with WIP-expressing constructs(16), indicating that WIP is required to maintain intracellular WASP levels. Interestingly, CR16 is only detectable in brain and testes, and the level of N-WASP protein, but not the transcript, is decreased in the testes and Sertoli cells of the CR16-knockout mice(11). These data support the idea that WIP is more abundantly expressed in hematopoietic tissues, where WASP is almost exclusively expressed and requires stabilization, while other proteins of the family such as CR16 are expressed in tissues where N-WASP degradation needs to be regulated.
This role of WIP has been conserved through evolution: human WASP suppresses the growth defect of the strain of Saccharomyces cerevisiae defective for las17, yeast orthologue to WASP, only in the presence of WIP. WIP mediates cortical localization of WASP as well as its stabilization in yeast cells(51).
WASP proteolisis by calpain contributes to the physiological turnover of podosomes, highly dynamic adhesion structures involved in cell motility and extracellular matrix degradation(52). Podosomes consist of a core of actin filaments forming a conical mass arising from the plasma membrane and protruding into the cytoplasm. The actin core also contains proteins involved in microfilament assembly (including WASP, cortactin and Arp2/3 complex) and is surrounded by a ring made up of vinculin, paxillin, talin, fimbrin, gelsolin, vimentin and other adaptor molecules that are linked to transmembrane adhesion molecules of the integrin family(53). Podosomes are naturally formed in migrating cells of monocytic/myeloid origin including DCs (Fig. 2) and osteoclasts, and are required for proper migration. Given that WIP was found in induced podosomes from endothelial cells(54) and had a role in fibroblast migration (Fig. 3), it was sensible to analyze WIP contribution to endogenous podosome assembly in DC and osteoclasts. Cultured murine WIP-null DC have a phenotype almost identical to WASP-null DC, that is, lack of podosomes and presence of evident focal adhesions accompanied by disorganized actin clumps(15) (Fig. 2). These data are understood in light of the diminished levels of WASP observed in DC devoid of WIP. The fact that lentiviral WASP overexpression in WIP-null DC does not recover podosome formation suggests that WIP is essential for correct WASP localization(15).
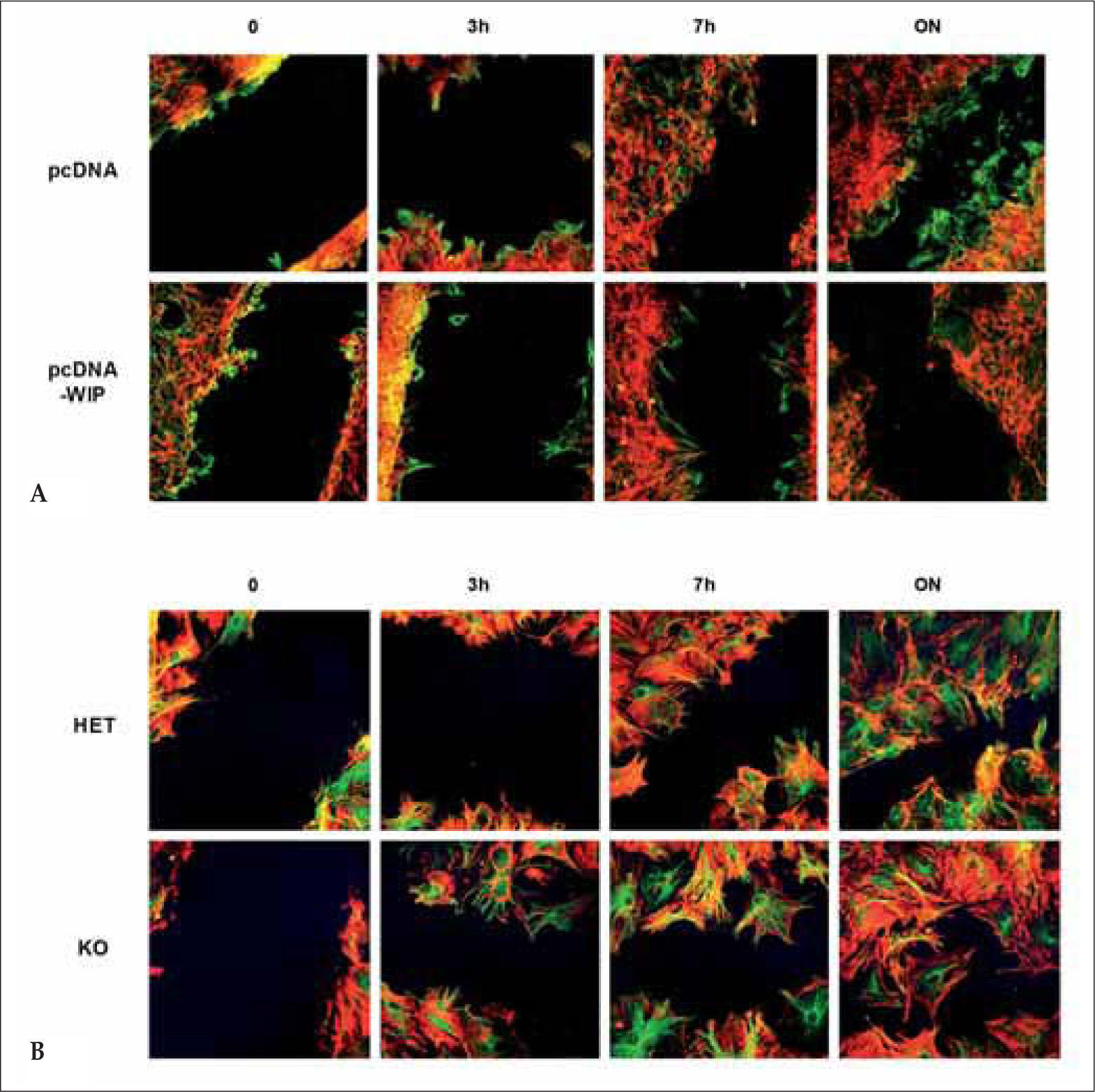
 WIP- overexpression decreases fibroblast migration. Transfected cells (pcDNA3 or pcDNA-WIP) were grown to confluency and scratched manually. Cells were incubated for the indicated times, fixed and stained (actin in red, tubulin in green). (B) WIP- deficiency increases fibroblasts migration. Murine fibroblast derived from control (HET) or WIP-deficient (KO) animals were grown to confluency on polylysine-coated coverslips and wounded by manual scratching. Cells were incubated and processed as described above.")
WIP participates in cellular migration. (A) WIP- overexpression decreases fibroblast migration. Transfected cells (pcDNA3 or pcDNA-WIP) were grown to confluency and scratched manually. Cells were incubated for the indicated times, fixed and stained (actin in red, tubulin in green). (B) WIP- deficiency increases fibroblasts migration. Murine fibroblast derived from control (HET) or WIP-deficient (KO) animals were grown to confluency on polylysine-coated coverslips and wounded by manual scratching. Cells were incubated and processed as described above.
Mature bone-resorbing murine osteoclasts are formed from fusion of mononucleated precursors derived from spleen or bone marrow. During maturation of cultured osteoclasts seeded onto non-mineralised substrates, podosomes undergo major reorganization(55): individual dynamic podosomes transform to 2-dimensional arrays (clusters of podosomes) and then reorganise into highly dynamic and short lived podosome rings to later form stable belts where podosomes are interconnected by an actin network(55). This podosome belt located at the cell periphery in mature osteoclasts is composed of two different actin-rich structures, the actin cloud and the podosome core. These two structures participate in cell adhesion and fulfill complementary roles since diffuse actin cloud contributes to cell contraction and podosome cores reinforce cell adhesion.
The requirement for WIP and several regulators of actin polymerization such as Arp2/3 complex, cortactin and WASP in podosome assembly in osteoclasts has been described. In mature murine osteoclasts the podosome core contained in the belt includes WIP, WASP, Arp3, cortactin and the non-integrin receptor CD44(56). WIP deficiency prevents podosome assembly while the formation of the actin cloud remains intact. The lack of podosomes is reversed by exogenous expression of WIP, supporting its crucial involvement in the process. The cloud also contains polymerized actin and adhesion-related molecules such as β3 integrin, paxillin and vinculin. In wild type murine osteoclasts, activation of β3 integrin by vitronectin (an αvβ3 ligand) leads to actin belt formation. In contrast, WIP-deficient osteoclasts plated on vitronectin are devoid of podosomes. Interestingly, activation of CD44 by osteopontin, collagen I, hyaluronic acid or activating anti-CD44 antibody favors podosome formation independently of WIP expression, suggesting the existence of different activation pathways for podosome formation involving different subsets of proteins.
Regulated cell migration is not only required for an efficient activation of the immune system but also for the correct development of multicellular organisms. Caenorhabditis elegans has often been used as a model for the study of hypodermal cell migration during morphogenesis in a process known as ventral closure. Arp2/3 complex, WSP-1 (WASP ortholog) and WIP-1 are identified players in this process. RNA interference (RNAi) treatment for either wsp-1 or wip-1 results in embryonic lethality with morphologic defects during ventral closure(57).
INFLAMMATIONThe initial influx of infiltrating leukocytes into sites of acute infection mainly relays on neutrophils. Within 16 h of an inflammatory reaction, neutrophils are replaced by monocytes/macrophages as the predominant infiltrating population(58). Chemotactic migration and accumulation of macrophages is critical for the subsequent recruitment of leukocytes to inflammed tissues since macrophages play a central role in the inflammatory process, releasing cytokines that control key events in the initiation, resolution, and repair processes of inflammation(58). The pathological accumulation of macrophages contributes to disease progression, such as chronic inflammatory diseases and cancer.
One key mechanism for the successful resolution of inflammation is the orchestrated clearance of apoptotic inflammatory cells by phagocytes (e.g., macrophages and dendritic cells)(59). Phagocytosis is also a first-line host defense mechanism against infection involving the ingestion and digestion of foreign materials such as bacteria. Phagocytes form an actinbased membrane structure called phagocytic cup at the plasma membrane whose formation is impaired in phagocytes from WAS patients. WASP and WIP form a complex at the phagocytic cup that plays a critical role in its generation. Impaired phagocytosis may cause defective clearance of apoptotic and dead cells, leaving cell debris available to resident dendritic cells. Presentation by DCs of self-antigens to the immune system in an inflammatory context could result in autoimmunity. This could be the underlying mechanism explaining the autoimmunity observed in mice lacking WIP. WIP-deficient mice display a progressive immunological disorder that resembles WAS in terms of inflammation and autoimmunity and ultimately results in the premature death of the animals(60). Only 10% of WIP−/− mice reach the age of ten months while the vast majority die between four and ten months of age (our unpublished observations). The absence of WIP produces progressive hematological alterations including granulocytosis and severe lymphopenia as well as an augmented number of segmented cells and a diminished number of bone marrow erythrocytes and lymphocytes.
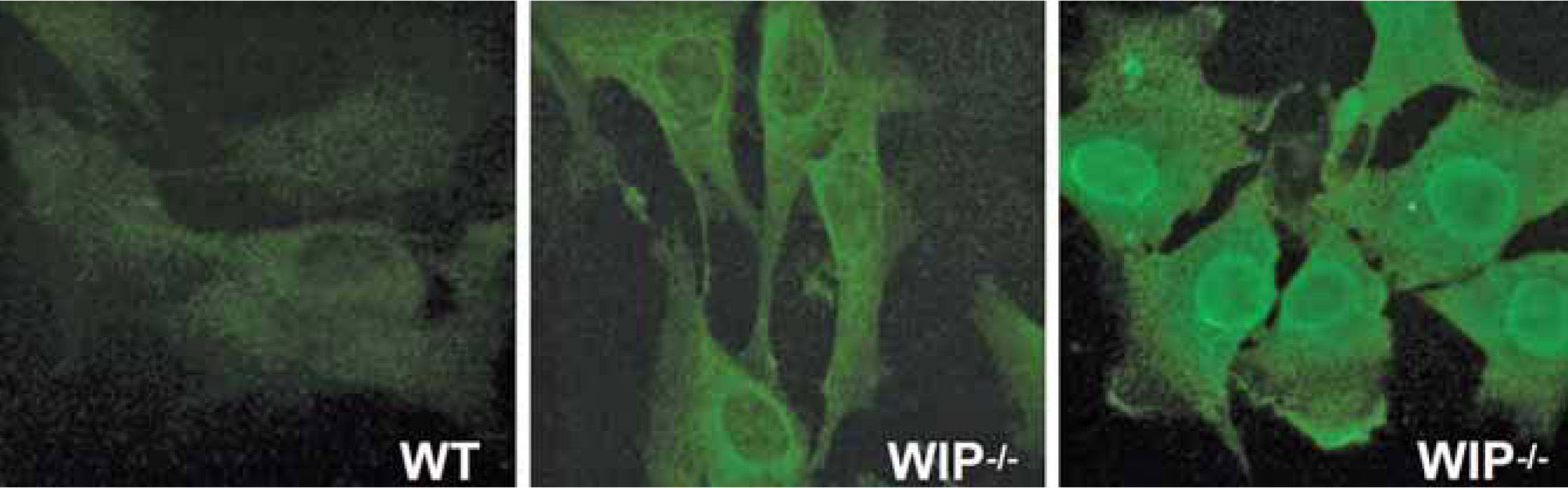
Progressive splenomegaly (reaching up to five times the size of control spleens) is accompanied by an increase in red pulp and a decrease in white pulp and marginal zone. The B lymphocyte (i.e. B220+) population is markedly reduced in the bone marrow, spleen, lymph nodes and Peyer's patches of WIP deficient mice. The most prominent pathological alterations associated with inflammation are observed in the intestine, lung and kidneys, where severe granulocyte and macrophage infiltration occurs, leading to ulcerative colitis, interstitial pneumonitis and glomerular nephropathy. The autoimmune origin of the pathological features observed is substantiated by the presence of autoantibodies in the serum of all WIP−/− mice analyzed (Fig. 4).
 and WIP−/−mice (WIP−/−) were diluted (1/100) and tested for autoantibodies using NIH3T3 murine fibroblasts as substrates for immunofluorescence microscopy. All WIP+ and none of the WT mice tested developed autoantibodies (n=3). Fluorescence location points to the nuclear, cytoplasmic or cytoskeletal origin of the antigens. All images were taken at identical exposure times.")
WIP deficiency induces autoimmunity. Sera from wild type mice (WT) and WIP−/−mice (WIP−/−) were diluted (1/100) and tested for autoantibodies using NIH3T3 murine fibroblasts as substrates for immunofluorescence microscopy. All WIP+ and none of the WT mice tested developed autoantibodies (n=3). Fluorescence location points to the nuclear, cytoplasmic or cytoskeletal origin of the antigens. All images were taken at identical exposure times.
WIP binds WASP, G-actin as well as F-actin and stabilizes microfilaments. Therefore WIP contributes to the regulation of cellular functions relying on reorganization of actin-rich structures such as adhesion, activation, migration, phagocytosis, endocytosis… WIP activity in the regulation of cellular activity is also mediated through its function as a chaperone for signaling molecules and as a protein shuttle to areas where cytoarchitecture remodeling is required.
WIP is very widely expressed but specially enriched in cells from the immune system. WIP deficiency, similarly to what is seen when deregulation of inflammation steps such as migration or adhesion occurs, leads to pathological situations associated to inflammation. Very recently, WIP deficiency has been linked to the development of autoimmunity in a mouse model. Further research is needed to establish the detailed molecular mechanism underlying the described phenotype. Most probably future data will support a similar role for WIP in human pathologies and the complete understanding of its role will contribute to the development of new therapies.
ACKNOWLEDGEMENTSThis work was supported by grants from the Spanish Ministry of Education and Science (BFU2007-64144) and CSIC-CAM (CCG07SAL-1967) to I.M.A. I. Bañón-Rodríguez is supported by a pre-doctoral contract from Consejería de Educación de la Comunidad de Madrid and Fondo Social Europeo (FSE). A. Franco is a recipient of a fellowship from Spanish Ministry of Education and Science (FPU).
DISCLOSURESThe authors declare no financial conflicts of interest.