










Polyacrylonitrile precursor fibers prepared using a solvent-free coagulation process were stabilized, carbonized, and physically activated by carbon dioxide into activated carbon fibers (ACFs). The activation temperature varied from 600 to 900°C while the activation time was 1h. Atomic force microscopy was used to observe the surface morphology, as well as the surface roughness of the ACFs. Higher pyrolysis temperature formed rougher surfaces, and increased the pore sizes. Meanwhile, Fourier transform infrared spectroscopy revealed more conversion of oxygen containing functional groups to carbonaceous materials as the activation temperature increased. Moreover, the microstructure properties were thoroughly characterized by the X-ray photoelectron spectroscopy (XPS) and X-ray diffraction (XRD) studies. XRD analysis showed that the activation of the ACFs shrank the ordered structure, reducing the D-spacing from 0.358 to 0.347nm for the fibers prepared at activation temperatures of 600 to 900°C. Meanwhile, XPS analysis concluded that that the oxygen containing functional groups were still retained even at high activation temperatures while the nitrogen containing functional groups were reduced during the high temperature activation in the CO2 atmosphere.
Activated carbon fibers (ACFs) can be prepared from various precursors namely polyacrylonitrile (PAN), coal, rayon, phenolic resins and pitches (Tsai, 1994). Different precursors will give different properties of ACFs and the pore texture of the ACFs significantly depends on the nature of the precursors (Chiang, Lee, & Lee, 2007). Among ACFs based on many different precursors, PAN based ACFs have attracted much attention of many researchers due to its high adsorption performance when compared to other counterparts. However, the organic solvents such as dimethylformamide (DMF) and dimethylacetamide (DMAc), which were often employed in the conventional coagulation bath in the fabrication of PAN fibers, could cause cancer after a long period of exposure due to its carcinogenic effects (Ismail, Rahman, Mustafa, & Matsuura, 2008; Yusof & Ismail, 2012a; Yusof, Ismail, Rana, & Matsuura, 2012). Therefore, in an attempt to reduce the hazardous effects, we investigated the properties of PAN/acrylamide (AM) fiber using a solvent-free coagulation bath in hollow fiber spinning of the polymer dope. It is known that the micro-structure of the ACFs finally obtained depends not only on the nature of the precursor but also on the conditions of activation that follows the precursor hollow fiber spinning, such as the activation temperature and the activation time (Sedghi, Farsani, & Shokuhfar, 2008). Nevertheless, there have been no detailed studies until now to know how the activation procedure affects the surface structure of PAN-based ACFs, particularly when the precursor PAN hollow fibers are spun in the solvent-free coagulation bath. It should be noted that the presence of pores on the rayon-based ACFs surface was recently confirmed using AFM (Wang et al., 2011; Zhao, He, Jiang, Li, & Li, 2007). It seems therefore very interesting to apply the AFM and other characterization methods to investigate the effect of activation conditions on the surface structures of the newly developed ACFs.
Therefore, the objectives of this study are to investigate the manipulation of the activation temperature toward the enhancement of micro porous structure of PAN/AM-based activated carbon fibers prepared using a solvent-free coagulation bath. In pursuit of this goal, the fibers were extensively characterized by means of atomic force microscopy (AFM), Fourier transform infrared (FTIR) spectroscopy, wide angle X-ray diffraction (XRD), and X-ray photoelectron spectroscopy (XPS). With the aid of these analysis methods, it is expected that this will be helpful for us to understand the relationship between the morphological behavior and chemical structure of PAN-ACFs prepared by different activation conditions. The results obtained from this study were also compared to the trends reported by previous studies.
2Experimental2.1Materials and fabrication of PAN fibersPolyacrylonitrile (PAN) in powder form was obtained from Aldrich, USA. Meanwhile, acrylamide (AM) purchased from Sigma–Aldrich, Germany was used as an additive. Dimethylformamide (DMF) purchased from Merck, Germany was employed as the solvent. The method to prepare uniform spinning dope of PAN and AM in DMF was reported earlier (Ismail et al., 2008; Yusof & Ismail, 2012a; Yusof et al., 2012). Eighteen weight percent of PAN/DMF solution was prepared, into which 5% of AM was added. The slurry was heated continuously at 70°C until a highly viscous dope solution became ready for the spinning process. Then, the homogeneous dope was degassed in an ultrasonic bath (Branson Ultrasonics) for 24h to remove the gas bubbles present in the dope. The dry-jet-wet spinning technique was used to produce PAN fibers. The high interest regarding environmental issues has resulted in the development of PAN fiber fabrication in a solvent-free coagulation process using 100% tap water in the coagulation bath (Ismail et al., 2008; Yusof & Ismail, 2012a; Yusof et al., 2012). The spinning conditions used in this study are listed elsewhere (Yusof & Ismail, 2012a). The AM/PAN fibers have a round shape cross-section with a fiber diameter around 100μm.
2.2Pyrolysis processThe stabilization process was carried out under tension in an air flow condition by heating at a rate of 2°C/min until 275°C, before constant heating carried on for 30min. This is an important step in preparing the fibers so that they can withstand higher temperatures without decomposing during the carbonization treatment by further orienting and then cross-linking the molecules (Sedghi et al., 2008; Yusof & Ismail, 2012b). The quality of the resulting activated carbon fibers depends strongly upon the degree of stabilization (Yusof & Ismail, 2012b).
The carbonization process was carried out without tension in a high-purity nitrogen stream with a flow rate of 0.2L/min at a heating rate of 3°C/min until 600°C was reached. Then the temperature was kept at 600°C for 30min, which is known as the soaking time. The activation step of the fibers was done with the introduction of carbon dioxide gas with a flow rate of 0.2L/min at a heating rate of 5°C/min and at various activation temperatures of 700, 800 and 900°C for 1h of activation time. The fibers made at the activation temperature of 700, 800 and 900°C are named as ACF 700, ACF 800, and ACF 900, respectively. For the case of ACF 600, the fiber is made at the 600°C activation temperature for 1h in the carbon dioxide gas atmosphere. It is noted that neither tension nor load was applied to the fiber during this process. The details of the heat treatment protocol can be illustrated in Figure 1.
2.3Characterization2.3.1Observation of surface and surface pores by the atomic force microscopy (AFM)
The surface morphology and roughness (Ra) of the ACFs were observed by atomic force microscopy (AFM). The samples were washed in ethanol to remove all traces of glycerol. Small pieces were cut from each fiber, fixed onto magnetic disks by using double side adhesive tape and then attached to a magnetic sample holder, located on top of the scanner tube. In order to analyze the fibers internal surface, the fibers were cut longitudinally by means of a sharp razor (Hallmark Blades Ltd., Sheffield, UK). Samples intended for observation of the internal surface were sectioned on the bias so that their cylindrical shape was retained and not strained during observation.
The laser beam of the AFM was focused on a preselected spot of the surface prior to the engagement of the cantilever. AFM was operating in a tapping mode at the surface of the ACFs. Surface scanning was done at an ambient temperature under the air environment using a NanoScope III AFM equipped with a 1533D scanner (Digital Instruments, Santa Barbara, CA). The Ra was measured by visual inspection of the line profile from different areas of the carbon fiber using the NanoScope software.
2.3.2Fourier transform infrared (FTIR) spectroscopyFourier transform infrared (FTIR) spectroscopy was used to observe the presence of functional groups and chemical structure transformation of PAN/AM fibers prepared under different heat treatment stages of the fiber. The FTIR Nicolet Magna-IR 560, with potassium bromide (KBr) pellet, was used to identify and classify the chemical structure transformation of PAN/AM fibers prepared under different heat treatment stages. An IR source at 45° incident angle was employed. Both the top and bottom layer of the asymmetric fiber surface samples were mounted with facing the crystal surface. The spectra were measured at room temperature in transmittance mode over a wave number range of 4000–600cm−1 at a resolution of 4cm−1 and averaged over 16 scans per sample.
2.3.3X-ray diffraction (XRD)X-ray Diffraction (XRD) Rigaku Rint 1200 with nickel-filtered Cu Kα radiation (λ=0.154056nm) at 40kV and 20mA was performed on the PAN fibers in order to observe its crystalline related properties. The diffractogram was scanned with a scanning rate of 2°min−1 in a 2θ range of 5–40° at room temperature. The data was corrected for Lorentz and polarization effects, and finally normalized to a convenient standard area. The crystallinity of the samples was calculated according to this Eq. (1):
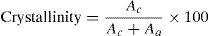
The interplanar distance (D-spacing) measurement can be obtained from the peak which appeared in the XRD pattern. The D-spacing of the fibers was calculated by the well-known Bragg equation, Eq. (2) as follows
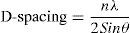
The stacking size (Lc) was calculated by the Scherrer formula, Eq. (3):
where k is the apparatus constant (=1), λ=0.154056nm and β is the half-value width in the radian of X-ray diffraction intensity vs. 2θ curve.2.3.4X-ray photoelectron spectroscopy (XPS)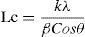
The elemental composition at the surface of the fiber was determined by X-ray photoelectron spectroscopy (XPS, Kratos Axis HS X-ray photoelectron spectrometer, Manchester, UK). Each random fiber was cut into samples of 1cm from the carbon fiber. Monochromatized Al Kα X-radiation was used for excitation and a 180° hemispherical analyzer with a three channel detector was employed. The X-ray gun was operated at 15kV and 20mA. The pressure in the analyzer chamber was 1.33×10−4–1.33×10−5Pa. The size of the analyzed area was about 1mm2. All of the carbon fiber surfaces were analyzed for specific element content at a take-off angle of 0° which corresponded to the X-ray penetration depth of 6.3nm. The angle between the normal to the sample and detector is the take-off angle (θ). At θ=0° (i.e. sample was perpendicular to the detector), the maximum sampling depth is achieved which is 6.3nm.
3Results and discussion3.1Morphological structure propertiesThe fiber surface topographies of top view and 3-D images were observed using AFM. The high peaks that are seen as bright regions in the AFM images characterize the nodules, while the pores are seen as dark depressions. As shown in Figure 2, the AFM images revealed that the surfaces of the precursor fiber had nodule like structures and the nodule aggregate was formed at the surface of the fiber. As can be appreciated, there is a gradual transformation of the precursor fiber depicted in Figure 2 into the final activated carbon fiber as shown in Figure 3.


Initially, the precursor fiber presents a structure made up of long fibrils, approximately parallel to the fiber axis as a reflection of its molecular conformation shown in Figure 2. Upon the activation, the fibrils were truncated at several points, losing their long range (along the fiber axis) cohesiveness. Upon pyrolysis at 900°C, the anisotropy of the fiber had disappeared completely and its morphology consisted of more or less rounded or elongated features arranged in a disorganized way. After activation, the characteristics of ACFs are significantly altered with the grooves and striations on the surface vanished while lots of pores emerged. The changes are mainly caused by activation (Wang et al., 2011). Similar morphological structure properties were also observed for ACFs previously studied by researchers (Wang et al., 2011; Brasquet, Rousseau, Estrade-Szwarckopf, & Le Cloirec, 2000).
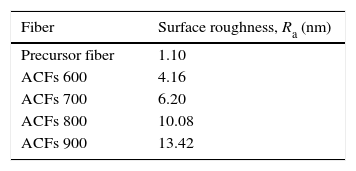
The images also further indicated that the surface of the fiber becomes rougher by the increment of activation temperature as proven by the AFM images in Figure 3. In line with the physical observation, the surface roughness of ACFs was also increased after activation. Thus, referring to the data tabulated in Table 1, the mean surface roughness, Ra of the activated carbon fibers are increased from 4.16 to 13.42nm (increment of Ra more than 3 times) with the increased activation temperature from 600 to 900°C.
3.2Chemical structure propertiesFTIR analysis is a well known and useful method for comparing qualitatively either vibrating absorption spectra of fibers or relative intensities of the respective band (Lv, Ge, & Chen, 2009; Ryu, Rong, Zheng, Wang, & Zhang, 2002; Yusof & Ismail, 2012a). Figure 4 shows the chemical structure properties of activated carbon fibers prepared under different activation temperatures and heated in CO2 gas stream for 60min soaking time. There are two main broad bands in the spectra as shown in Figure 4. The band at 1220cm−1 is partly associated with CO stretching and OH bending modes in the functional group (Ryu et al., 2002).

Meanwhile, the band in the region of 1560cm−1 is assigned to an aromatic ring stretching mode or a highly conjugated hydrogen bonded CO, analogous to the structure of acetyl acetone due to the physical activation by CO2 gas (Figueiredo, Pereira, Fritas, & Órfão, 1999; Pradham & Sandle, 1999). As observed in Figure 4, for ACFs with higher activation temperatures (700–900°C), it can be noticed that there is a decrease in the intensity of the bands at 1220cm−1 and 1560cm−1 with the increase of the activation temperature from 600 to 900°C. All of the spectra were somewhat similar, and the main difference was the low intensity of oxygen functional groups as the activation temperature was increased to 900°C indicating that more carbonaceous materials were formed at a higher temperature.
3.3Microstructure propertiesXRD is a useful tool for understanding the organization of carbon at the molecular level and provides qualitative and comparative assessment of the degree of packing of microstructures. The most relevant peaks when examining polymer-based carbon fibers are the (002) and (10) peaks as stated by previous researchers (Lu & Zheng, 2001; Ryu et al., 2002; Yu, Bai, Wang, Xu, & Guo, 2007). In this research, the XRD spectra for the ACFs treated at different activation temperature during activation step are shown in Figure 5. Two broad peaks at low diffraction angles are observed to correspond to the diffraction angles 2θ at 25.5–26.5° and 36–43.30°, which are assigned to the 002 plane and 10 plane (overlapped 100 and 101) of disordered micro-graphite stacking, respectively (Ryu et al., 2002; Short & Walker, 1963). The (002) peak is commonly used to determine the crystal length, which can be attributed to the turbostratic structure with randomly oriented graphitic carbon layers while the (10) peak is related to the distance between carbon atoms on the same plane (Ryu et al., 2002).

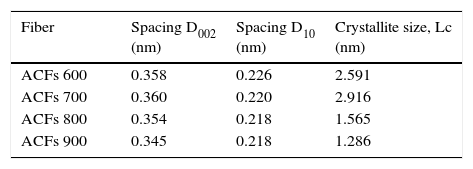
All of the ACFs samples showed wide peaks indicated the degree of amorphousness and appeared very close to the corresponding value for graphite of 0.335nm with a diffraction peak appearing at 26.58° (Sutasinpromprae, Jitjaicham, Nithitanakul, Meechaisue, & Supaphol, 2006). It proves that the physical activation process by CO2 gas stream toward the PAN/AM fibers prepared from the solvent-free coagulation process is a success. Concerning the plane 002, the diffraction peak appears between 2-θ degrees of 25.50–26.50° for the prepared ACFs under activation temperature of 600–900°C, corresponding respectively to a d-spacing of around 0.3578–0.3465nm. From the D002 spacing data tabulated in Table 2, it demonstrates that as the activation time increases, the graphite like layer spacing (d-spacing) of fibers declines from 0.3578 to 0.3465nm. The decrease of the d-spacing as the activation temperature increases was also observed for ACFs prepared from physical activation previously studied by Su, Ko, and Lin (2008).
Based on XRD spectra on the ACFs prepared at different activation conditions, it can be concluded that the graphite sheets have been constructed in carbon structure during the activation step. The activation of the ACFs shrank the ordered structure, reducing the d-spacing from, 0.358 to 0.347nm, which corresponds to 3.5% contraction, for the fibers prepared at activation temperature of 600–900°C. Meanwhile, another peak correlated to the graphite (10) plane, D10 spacing, represents the repeated aromatic ring in the graphitic structure between 2θ=43–45.5° was observed in all samples of the activated carbon fibers as tabulated in Table 2.
Referring to the crystallite size data in Table 2, all of the ACFs samples prepared contain disordered graphite micro-crystallites, with inter-crystallite and intra-crystallite voids forming the pores with the crystallite size of ACFs ranging from 1.286 to 2.916nm. From the XRD results, the crystal size decreases with the increase of the activation temperature. Similar findings were reported by previous researchers (Lu & Zheng, 2001; Ryu et al., 2002). ACF 900 has the smallest crystallite size (1.286nm) and the shortest D002 spacing was observed at 0.345nm. A small crystallite size and short range ordering between crystallites provides the prerequisites for the realization of high surface area activated carbon fibers (Ryu et al., 2002). Thus, a short conclusion can be made that 900°C is the optimum activation temperature for the production of high performance adsorbent media.
Meanwhile, the chemical composition and binding characteristic at the surface of the fiber was determined by X-ray photoelectron spectroscopy investigation. The X-ray photoelectron spectroscopy spectra of the ACFs samples contain three distinct major peaks due to carbon, nitrogen and oxygen. In order to clarify the functionalities and their variation with the activation temperature, curve fitting procedures on the C1S, O1S and N1S spectra were investigated. Referring to Figure 6(a–d), it can be concluded that all of the prepared ACFs samples show an asymmetrical shape for the C1S spectra extending from 283 to 288eV.

This chemical shift toward higher binding energies indicates that there are various carbon-based functional groups on the surface of these ACFs. A major peak at 284±0.5eV is attributed to CC/CH of carbidic/graphitic carbon (Biniak, Szymański, Siedlewski, & Świątkowski, 1997; Jansen & van Bekum, 1995). In addition, a series of chemically shifted peaks is observed at the high-binding energy side. Several minor peaks are generally observed, which occur at binding energies at about 286±0.5eV, 287±0.5eV and 289±0.5eV, respectively. They can be assigned to CO (ether/hydroxyl) and/or CN, CO (carbonyl), and OCO (carboxyl) functionalities respectively appeared upon the heat treatment (Biniak et al., 1997; Wang, Hong, & Zhu, 1996).
The typical O1S spectra in Figure 7(a–d) appear to contain mainly three chemically shifted peaks. The main peak at a binding energy of about 531±0.5eV is attributed to CO groups (ketone, lactone, carbonyl). While the peak at 533±0.5eV is probably due to CO in esters, amides, carboxylic anhydrides and oxygen atoms in hydroxyls or ethers, while the other peak at 535±0.5eV is tentatively assigned to chemisorbed oxygen and/or water. Thus, based on the C1S and O1S spectrum analyses, it can be interfered that there are several forms of carbon-oxygen functionalities present on the surface of the fiber.

Meanwhile, the N1S spectra in Figure 8(a–d) indicates that not only does nitrogen exist in various functionalities on the surfaces, but these nitrogen functionalities vary to some extent from activation temperature of 600–900°C (Wang et al., 1996). The N1S spectra contain two major chemically shifted peaks: isonitriles compound (398±0.5eV) and aromatic amine (399±0.5eV) (Ryu et al., 2002). Other assignments of peaks 399.0–400.0eV could be attributed to amide functionality due to the addition of acrylamide in the precursor PAN/AM fibers, and the peak at 400–401eV were credited to aliphatic amine, while the peak around 400.9–401.7eV was probably due to protonated primary amine (NH3+) (Wang et al., 1996). From the N1S spectra, it can also be observed that the intensity of the lower binding energy peak gradually decreased when subjected to the higher activation temperature. This suggested that the elimination of aromatic amine occurred during these heat treatment activities.
4Conclusions
The AFM micrographs analysis shows that porosity is increased together with the loss of volatile components through the activation process. Meanwhile, the success of physical activation of carbonized AM/PAN fiber using CO2 gas at 900°C is proved by FTIR, XRD, and XPS analysis. The intensity of oxygen functional groups is lowered as the activation temperature was increased to 900°C indicating that more carbonaceous materials were formed at a higher temperature. The activation of the ACFs shrank the ordered structure, reducing the d-spacing from, 0.358 to 0.347nm with an increase of temperature. Thus, based on the C1S and O1S spectrum analyses, it can be interfered that there are several forms of carbon-oxygen functionalities present on the surface of the fiber. The N1s results suggested that the elimination of aromatic amine occurred during these heat treatment activities. These observations infer that nitrogen originating from PAN, AM and DMF decreases but oxygen containing functional groups remain, while hollow fiber is heated in CO2 atmosphere. In the meantime, d-spacing decreases leading to more compact structure. It is also proved that the precursor fibers prepared via environmental friendly solvent-free coagulation processes that the micro-porous structure confirmed its potential to be used as an adsorbent media for gas separation in the near future.
Conflict of interestThe authors have no conflicts of interest to declare.
The authors would like to acknowledge the financial support from Universiti Teknologi Malaysia under Grant Q.J130000.2542.05H46 and Q.J130000.2546.12H54. The authors would also like to acknowledge the technical and management support from Research Management Centre (RMC), Universiti Teknologi Malaysia.
Peer Review under the responsibility of Universidad Nacional Autónoma de México.