





La hiperplasia suprarrenal congénita se debe en un 95% de los casos a la deficiencia del esteroide 21-hidroxilasa (21-OHD). Esta enfermedad autosómica recesiva presenta un amplio espectro de formas clínicas, desde las clásicas neonatales, que en su variante perdedora de sal pueden comprometer la vida de estos niños, hasta formas atenuadas denominadas no clásicas (21-OHD-NC), en que los síntomas aparecen en la etapa pediátrica y/o adulta, e incluso formas crípticas u oligosintomáticas. Es una enfermedad común en sus formas leves, especialmente en el área mediterránea1, y no infrecuente en sus formas graves (frecuencia de portadores de mutación grave de 1:50-1:60), en las que cabe la posibilidad de plantear un tratamiento prenatal que previene la virilización de los fetos femeninos afectados1-3. Existe una estrecha correlación genotipo/fenotipo y algunos autores4 han señalado que la genotipificación de mutaciones puede ser relevante para la categorización de estos pacientes, ya que la expresividad/gravedad clínica podría estar condicionada por el estrés, el sexo del paciente y otros factores ambientales.
El cribado básico de 10 mutaciones puntuales (P30L, 655AoC * G, deleción 8pb, I172N, I236N/V237E/M239K, V281L, 306insT, Q318X, R356W, P453S), deleciones y conversiones permite caracterizar un gran número de los alelos deficientes. Hay un seudogén homólogo, duplicado en tándem, que presenta las alteraciones puntuales mencionadas y que, por procesos de recombinación asimétrica y conversión génica, puede originar los alelos deficientes1,5. Este mecanismo especial de producción de las mutaciones, junto con una diseminación preferente de algunos alelos mutados1,5, contribuye a la recurrencia y elevada frecuencia de éstos. El porcentaje de alelos 21-OHD caracterizados tras el cribado básico de estas mutaciones recurrentes en nuestra población es del 93% para las formas clásicas y algo menor para las no clásicas (85%)6,7.
El pequeño porcentaje restante de alelos 21-OHD clásicos presenta mutaciones producidas por los mecanismos tradicionales y su caracterización se realiza mediante la secuenciación completa del gen. Estas mutaciones no frecuentes o raras se encuentran generalmente en heterocigosis compuesta con las mutaciones antes mencionadas. Se considera que estas variantes raras son causantes de la deficiencia cuando suponen cambios graves que afectan a la fase de lectura, dan lugar a la aparición de codones de parada o a cambios de aminoácido no conservadores que afectan a regiones conservadas y/o a la funcionalidad de la enzima (estudios in vitro). Estas nuevas mutaciones también se heredan y se encuentran en los progenitores; aunque ya no son tan frecuentes como las anteriores, puede haber cierta recurrencia de las mismas en distintas poblaciones1,4,5. En ocasiones estas mutaciones raras pueden encontrarse en homocigosis en pacientes que resultan negativos en el cribado básico8.
La hiperplasia suprarrenal congénita por 21-OHD-NC se debe a heterocigosis u homocigosis para mutaciones leves o a heterocigosis compuesta de mutación leve y grave, situación que se da en más de un tercio de estos pacientes7,9, incluso en las formas oligosintomáticas10. Cuando se da esta situación, existe un riesgo superior de que en la descendencia pueda aparecer un caso afectado de la forma clásica1.
Inicialmente las formas no clásicas se identificaron en mujeres adultas en el contexto de hirsutismo y/o amenorrea. Su diagnóstico en pacientes pediátricos es más reciente. Estas formas no clásicas no están bien definidas en el varón adulto, en el que se reconocen con menos frecuencia, y habitualmente se diagnostican cuando se realizan estudios familiares a partir de una mujer afectada.
Presentamos el casos de 2 hermanos, mujer y varón, ambos clínicamente sintomáticos, en quienes se detectó una heterocigosis compuesta con mutación grave que se describe por primera vez, y discutimos la importancia de su valoración clínica y de la realización del consejo genético.
Pacientes y método
Caso índice
Mujer de 19 años, sin antecedentes personales de interés, que consultó por amenorrea secundaria e hirsutismo, sin síntomas de virilización. En la anamnesis familiar refería hirsutismo en el padre y un hermano (tabla 1). En la exploración física presentaba una estatura de 150 cm, índice de masa corporal de 24,4, laringe algo prominente, mamas hipoplásicas y discreta hiperplasia clitorídea. La valoración del mapa de Ferriman fue de 10 puntos, con predominio en los muslos y mentón. Los resultados del estudio bioquímico se recogen en la tabla 1.

Caso 2: hermano afectado
Varón de 25 años, con antecedentes de pubarquia a los 8 años y pubertad a los 13. Había consultado a los 16 años por alopecia y refería hirsutismo de siempre, con aparición de vello abundante en la espalda a los 22 años. En la exploración física se observó alopecia frontoparietal moderada-grave, con una valoración de V-VI según la escala de Hamilton-Norwood11, además de hirsutismo en brazos, piernas, tronco y área superior de la espalda. La estatura era de 165 cm. Los parámetros bioquímicos analizados se recogen en la tabla 1.
Dos observadores independientes realizaron la valoración de la gravedad del hirsutismo mediante la escala semicuantitativa de Ferriman Gallwey. La valoración de la alopecia se realizó mediante comparación con los tipos de la escala de Hamilton-Norwood11 por 2 observadores independientes.
Estudio molecular
Se extrajo ADN a partir de leucocitos de sangre periférica de ambos pacientes, padres y hermanos y se procedió a la amplificación específica del gen CYP21A2 según la técnica descrita por Ezquieta et al6. El análisis directo del gen se realizó en 2 etapas: a) un primer cribado de las mutaciones puntuales recurrentes (P30L, 655G, I172N, triple mutación del exón 6, V281L, 306insT, Q318X, R356W, P453S) mediante reacción en cadena de la polimerasa, hibridación específica de alelo y separación en geles nativos de acrilamida para la deleción 8pb del exón 3, como se ha descrito en otra publicación6, b) un estudio complementario de marcadores tipo microsatélite5,8 de la región HLA D6S273, D6S439 y polimorfismo intrónico del gen TNF para evidenciar si ambos hermanos compartían los marcadores de los alelos 21-OH deficientes, y c) el estudio se completó con la secuenciación de las regiones codificantes y barreras intrón-exón, así como 50 pares de base de las regiones 5' (promotor) y 3' (región no traducida) utilizando dideoxinucleótidos fluorescentes y ABIPrism 3100 (Línea Instrumental de Secuenciación del Hospital Gregorio Marañón), como han descrito Ezquieta et al8.
Determinaciones hormonales
Se extrajeron muestras basales de 17-hidroxiprogesterona (17-OH-P), testosterona, delta 4-androstenediona, sulfato de dehidroepiandrosterona, hormonas luteinizante (LH) y foliculostimulante (FSH) en ayunas entre las 8.00 y 9.00 h, y de 17-OH-P 60 min después de una inyección intravenosa de 250 µg de tetracosáctido (Synacthen, Novartis Farm). También se determinaron la aldosterona y la renina plasmáticas y, en el caso índice, la LH y FSH 30 y 60 min tras la inyección intravenosa de 100 µg de hormona liberadora de gonadotropinas (Luforán, Serono).
La 17-OH-P se midió en suero por radioinmunoanálisis (RIA) competitivo con doble anticuerpo (Pantex, Division Bio-Annalysis). Los valores de referencia del Laboratorio de Hormonas del Hospital 12 de Octubre, que se indican al pie de la tabla 1, se relacionan bien con los suministrados por la casa comercial. El método detecta valores de 0,1 a 20 ng/ml. A partir del año 2004 se sustituyó el método por otro RIA también competitivo, pero con tubos recubiertos de anticuerpo (Immunotech, Beckman Coulter Company). Este método detecta de 0,1 a 50 ng/ml, y nuestros valores de referencia, que son sensiblemente más altos que los anteriores (tabla 1), se ajustan a los proporcionados por la casa comercial.
La testosterona, delta 4-androstenediona y el sulfato de dehidroepiandrosterona se analizaron en suero, por RIA competitivo, y la LH y FSH por un método de enzimoinmunoanálisis (Boehringer Mannheim). La aldosterona se determinó en plasma, en reposo, por RIA competitivo (DiaSorin), y la renina activa se midió en plasma mediante análisis inmunorradiométrico (Nichols Institute Diagnostics).
Resultados
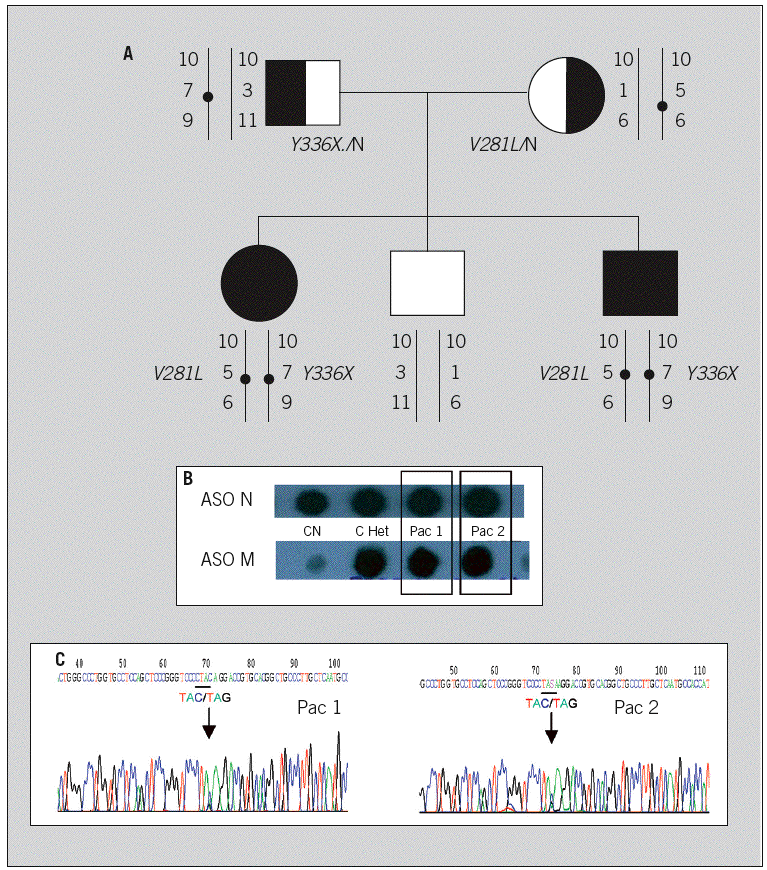
El árbol familiar se muestra en la figura 1A, en que se detalla la segregación de alelos en los análisis directo (mutaciones detectadas) e indirecto (alelos polimórficos de tipo microsatélite) en los distintos componentes de la familia. El cribado básico de mutaciones mostró que ambos hermanos portaban la mutación V281L en heterocigosis (fig. 1B) y fue negativo para el resto de mutaciones recurrentes, lo que indicaba una caracterización genotípica parcial. El análisis indirecto mediante marcadores tipo microsatélite en la región HLA (D6S273, marcador estrechamente ligado al gen, y 2 marcadores más alejados en 5' y 3', marcador intrónico del gen TNF y D6S439, respectivamente) mostró que ambos hermanos eran idénticos (fig. 1A). Ambos presentaban el marcador D6S273 de 140 pares de base, que en nuestra población se asocia con al alelo portador de la mutación puntual V281L. El estudio del complejo principal de histocompatibilidad, que se había realizado con anterioridad al estudio molecular (año 1992, Laboratorio de Inmunología del Hospital 12 de Octubre), había revelado que ambos hermanos eran idénticos y que en uno de los alelos compartidos portaban el marcador B14, relacionado con la mutación típica de formas no clásicas, V281L (detectada ahora de forma directa en los estudios realizados sobre el ADN).
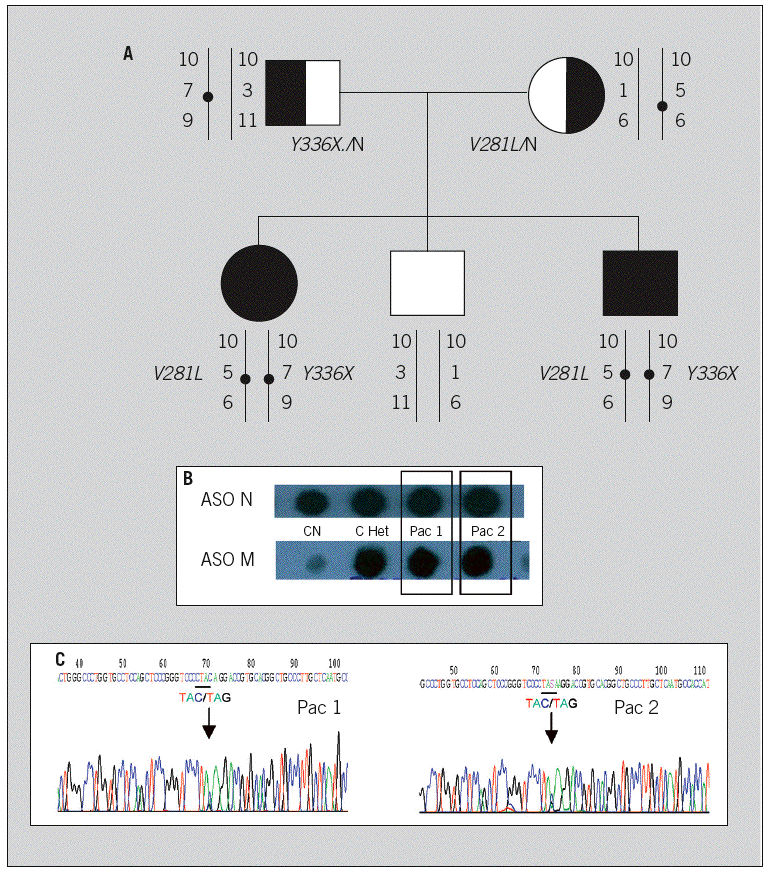
Fig. 1. Estudio molecular de los pacientes y familiares. Árbol familiar (A), en que se recogen los resultado de los análisis, directo e indirecto, realizados sobre el ADN de padres, casos índice y hermano. El análisis directo correspondiente al cribado básico de las mutaciones recurrentes muestra que ambos presentan la mutación Val281Leu (B). La secuenciación complementaria (C) del exón 8 en ambos pacientes detecta el cambio C * G en heterocigosis en la posición 2055, que supone el cambio de triplete de la posición 336, que es una tirosina (TAC, Y), en el alelo normal y un codón de parada (TAG, X) en el alelo mutante.
El estudio complementario de secuenciación (fig. 1C) detectó en ambos hermanos, en el exón 8, codificante de una de las regiones del centro activo de la enzima, un cambio de base C * G que afecta al triplete codificante Tyr336 y que da lugar a la aparición de un codón de parada (Y336X). La lectura de la secuencia complementaria confirmó estos hallazgos. La mutación Y336X, al igual que la mutación V281L, se encontró en heterocigosis con la secuencia normal. También se detectaron otros cambios en la secuencia, que en estos casos correspondían a polimorfismos frecuentes del gen: P45, L248 y S268T. Los 2 primeros no ocasionan cambio de aminoácido, y el tercero, aunque en un primer momento, cuando comenzaron a caracterizarse las alteraciones puntuales del gen 21-OH, se describió como mutación, fue posteriormente documentado in vitro como variante que no afectaba a la actividad de la enzima12.
El estudio familiar molecular (fig. 1A) permitió documentar la segregación de alelos en los progenitores: el padre es portador de la nueva mutación Y336X y la madre presenta la mutación leve V281L, por lo que los pacientes son heterocigotos compuestos para dichas mutaciones. El estudio del hermano permitió conocer que era sano, no portador.
Discusión
La 21-OHD en su forma atenuada o no clásica constituye el trastorno hereditario autosómico recesivo más frecuente en la población1. La estimación de la frecuencia de la enfermedad varía, dependiendo de la etnia y área geográfica estudiada, entre un 0,1 y un 3-4%13. Estudios moleculares de cribado en recién nacidos de población australiana describieron una proporción del 5% de portadores de mutaciones del CYP21A2 asociadas a la forma clásica o no clásica (un 2,8 y un 2,2%, respectivamente)14. Otros estudios recientes, basados en población centroeuropea, encuentran una frecuencia de portadores de la deficiencia del 9,5% (un 5,5% para la forma clásica y un 4% para la no clásica)15. La enfermedad es más frecuente en judíos del este de Europa (1:27), hispanos (1:53), yugoslavos (1:63) e italianos (1:333). En cuanto a nuestra población, disponemos de datos orientativos recientes que proceden de la estimación de la frecuencia de portadores de la mutación puntual V281L (responsable de más de un 57% de los alelos en formas no clásicas) en muestras del cribado neonatal de la Comunidad de Madrid. De estos datos se deduce una frecuencia de 1:225 para la forma no clásica, en la que quedarían incluidas las formas crípticas16.
Se desconoce la verdadera prevalencia de la 21-OHD-NC obtenida a partir de casos afectados. Tampoco se conoce bien la proporción de individuos con hiperandrogenismo sintomático, ya que muchas mujeres no consultan y prácticamente ningún varón lo hace por síntomas relacionados con hiperandrogenismo. De hecho, en el estudio de Knochenhauer et al17, en que se concluye que los portadores de 21-OHD no presentan signos de exceso de andrógenos, se estima una frecuencia de hiperandrogenismo del 5-20% en población sana. En la edad pediátrica, la frecuencia de 21-OHD en niños con pubarquia precoz no es baja (un 5-10% según las series)18. Entre las mujeres que consultan por síntomas de hiperandrogenismo la frecuencia de la forma no clásica oscila, según la población estudiada, entre un 1,6 y un 9,5%19. Aunque la enfermedad se da por igual en ambos sexos, es llamativo el escaso número de varones (89 frente a 287 mujeres) en la amplia serie de pacientes diagnosticados en el estudio de New20. Esta desproporción es menor en población pediátrica (23 varones frente a 63 niñas)7, probablemente porque los síntomas que son determinantes en el diagnóstico aceleración de la edad ósea o pubarquia prematura aparecen también en el niño.
Los 2 hermanos presentados son heterocigotos compuestos para la mutación leve V281L y una mutación de codón de parada (Y336X), que no se había descrito hasta ahora, siendo el fenotipo resultante el de una forma «leve». Éste sería el fenotipo esperado, ya que, aunque las mutaciones de codón de parada dan lugar a proteínas truncadas y se consideran de carácter grave, el fenotipo viene determinado por el alelo más leve, en este caso, V281L1. Desde el punto de vista clínico, la mujer presenta un hiperandrogenismo grave, con amenorrea, signos de virilización y síndrome del ovario poliquístico. Ambos hermanos presentan descenso de las cifras de aldosterona, que podría correlacionarse con un déficit enzimático más grave, aunque en ningún caso virilizante simple o clásico. En cuanto a la clínica del varón, la familia refiere aparición temprana de vello pubiano (probablemente antes de los 8 años), pero no existen datos de curva de crecimiento ni de maduración ósea, ya que no se consultó por ello. Tampoco fue motivo de consulta el hirsutismo llamativo y progresivo en la espalda, aunque el paciente sí consultó por la alopecia de inicio prematuro, síntoma al que en la literatura médica no se ha prestado atención en los varones afectados de deficiencia de 21-OH.
Los varones afectados suelen presentarse como asintomáticos, si bien esto se debe a la dificultad para reconocer en ellos los signos de hiperandrogenismo, que, por otro lado, serán de más difícil reconocimiento a medida que aumenta la edad. Los varones afectados por acné quístico o alopecia suelen dirigirse al dermatólogo, quien habitualmente no realiza cribado de hiperandrogenismo, o ni siquiera solicitan consulta médica. La alopecia androgénica depende, entre otros factores reguladores, de las concentraciones de andrógenos circulantes y de su intensidad y tiempo de acción sobre el folículo pilosebáceo del cuero cabelludo. Por ello, el inicio de la alopecia antes de los 30 años debería investigarse como síntoma de hiperandrogenismo desde el punto de vista bioquímico. Los varones con 21-OHD-NC pueden también presentar infertilidad, oligoazoospermia o insuficiencia funcional de las células de Leydig, debido a la presencia de tejido ectópico adrenal localizado en los testículos. Dado que el exceso de andrógenos adrenales puede inhibir la secreción de gonadotropinas y reducir tanto la secreción de testosterona como la espermatogenia, estos últimos casos podrían beneficiarse del tratamiento precoz con esteroides21,22. No hay datos relativos al efecto del tratamiento esteroideo sobre el resto de síntomas de hiperfunción androgénica en el varón.
Es importante considerar que, aparte de los temas relacionados con el tratamiento, no debe olvidarse el consejo genético adecuado. Las formas clásicas neonatales, y en concreto la virilización de las niñas afectadas, puede prevenirse mediante tratamiento prenatal. Este tratamiento, para el que debe contarse con el consentimiento informado y que debe realizarse en hospitales de referencia con seguimiento adecuado, únicamente está indicado si hay riesgo de feto afectado de la forma clásica 1:4 (ambos progenitores portadores de mutación grave). A priori, el riesgo de portador 21-OHD en la población general es de 1:60 y, obviamente, en los familiares de casos índice del déficit es de la mitad y un cuarto según lo sean en primero o segundo grado, respectivamente, como en cualquier enfermedad monogénica autosómica recesiva. En las formas no clásicas, un tercio de los pacientes son heterocigotos compuestos con mutación grave7,9, y éstos tendrán un 50% de probabilidad de pasar esta mutación grave al feto. El análisis molecular es el único que puede documentar la existencia de mutación grave y, como señalan algunos autores23, constituye el único método preciso para detectar portadores. Aunque los valores de 17-OH-P tras estimulación con hormona adrenocorticotropa (ACTH) pueden estar elevados (los valores basales son siempre normales), esto sólo ocurre en el 50-75% de los casos según las series7,23; más importante aún, los valores de 17-OH-P en portadores no permiten discriminar si la mutación es leve o grave. Dada la frecuencia de portadores de la deficiencia, la posible coincidencia de una pareja portadora de una mutación grave ha de tenerse en cuenta para la descendencia.
Es importante reseñar que la elevada frecuencia de la mutación leve V281L en nuestro medio estaría «facilitando» la potencial expresión clínica de estas mutaciones graves cuando, al encontrarse en heterocigosis compuesta con los alelos leves, dieran lugar a formas no clásicas de la deficiencia. El estudio molecular de estos pacientes brindaría la oportunidad de documentar la existencia de la mutación grave y plantear la potencial indicación de tratamiento prenatal. En cuanto a los estudios en población general, como serían los de las parejas de estos pacientes o de los portadores de la mutación grave documentados en los estudios familiares, ha de tenerse en cuenta que el cribado de mutaciones puntuales debe complementarse con técnicas que permitan descartar deleciones y duplicaciones del gen, como hemos tenido ocasión de describir recientemente24. Por otro lado, en lo que al diagnóstico preimplantacional se refiere, queremos reseñar que hay que ser cauto en la aplicación de abordajes de análisis directo, como recientemente hemos señalado en el estudio citado. Los loci complejos, como el que nos ocupa, al someterse a procesos previos de amplificación «genérica» del ADN, pueden alterarse, de modo que los resultados del análisis directo podrían no reflejar la constitución de los alelos presentes en el embrión analizado.
En resumen, el caso que presentamos ilustra y subraya la importancia de no subestimar los posibles síntomas de hiperandrogenismo en el varón adulto, ya que pueden ser secundarios a una enfermedad oculta, infradiagnosticada, posiblemente subsidiaria de tratamiento específico y con implicaciones genéticas y de planificación familiar.
