




Información del artículo
Resumen
Texto completo
Bibliografía
Descargar PDF
Estadísticas
Tablas (5)
Fig. 1. Ruta metabólica de los neurotransmisores. Las enzimas implicadas en estas enfermedades aparecen en cursiva con su coenzima entre paréntesis. Los metabolitos intermediarios que se cuantifican para el diagnóstico aparecen en negrita. La deficiencia de TH cursa con: disminución de HVA y MHPG, y 5-HIAA y 5-HTP normales. La deficiencia de AADC cursa con: disminución de 5-HIAA, HVA y MHPG, y aumento de 5-HTP y 3-OMD. La deficiencia de DßH cursa con: aumento de HVA, disminución de MHPG, y 5-HIAA y 5-HTP normales. 3-OMD: 3-ortometildopa; 5-HTP: 5-hidroxitriptófano; MHPG: 4-hidroxi-3-metoxifenilglicol; 5-HIAA: ácido 5-hidroxiindolacético; HVA: ácido homovanílico; TH: tirosina hidroxilasa; AADC: descarboxilasa de aminoácidos aromáticos; DßH: dopamina betahidroxilasa; BH4: tetrahidrobiopterina; B6: vitamina B6; Cu: cobre.
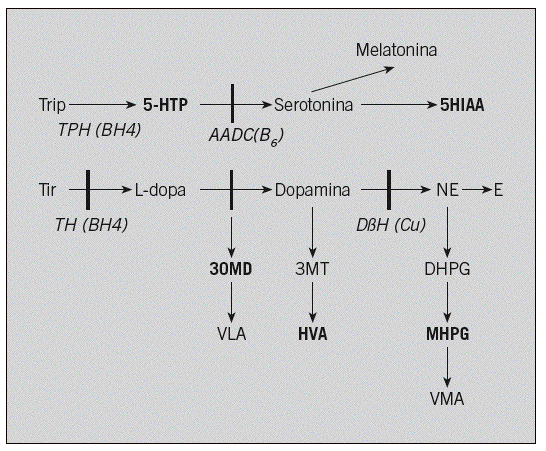
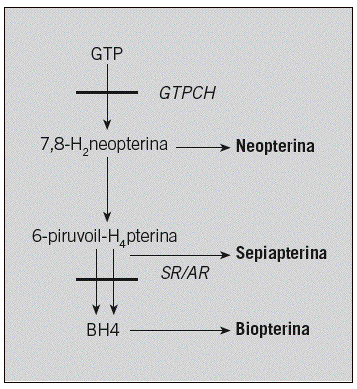
Fig. 2. Metabolismo de las pterinas. Las enzimas implicadas en estas enfermedades aparecen en cursiva y los metabolitos intermediarios que se cuantifican para el diagnóstico, en negrita. La deficiencia de GTP-CH1 cursa con: disminución de NP, BP y HVA, y 5-HIAA reducida o normal. La deficiencia de SR cursa con: aumento de BP y SP, disminución de HVA y 5-HIAA, y NP normal. GTP-CH1: guanosinatrifosfato ciclohidrolasa; SR: sepiapterina reductasa.


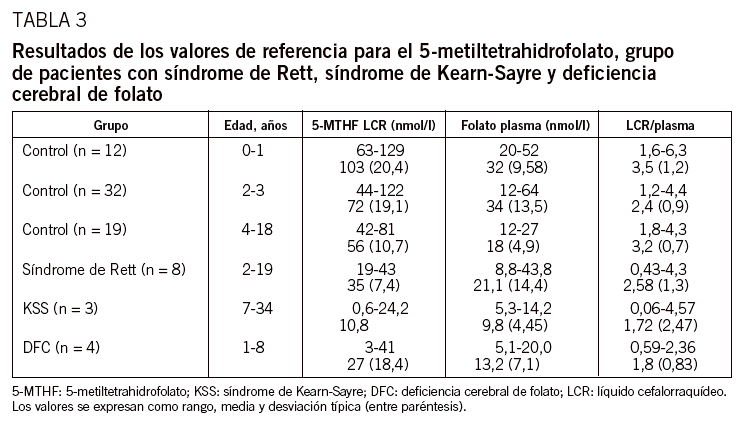
Mostrar másMostrar menos
Fundamento y objetivo: En la última década se ha descrito diferentes errores congénitos del metabolismo de los neurotransmisores (NT), en especial de las vías dopaminérgica y serotoninérgica y de las pterinas. También se ha descrito defectos primarios en el transporte de glucosa y 5-metiltetrahidrofolato (5-MTHF) a través de la barrera hematoencefálica, todos ellos enfermedades raras para cuyo diagnóstico es necesario el estudio en líquido cefalorraquídeo (LCR). Nuestro objetivo ha sido evaluar los resultados de la aplicación de un protocolo de análisis de LCR en España y Portugal durante 3 años en pacientes pediátricos con trastornos neurológicos de origen desconocido. Pacientes y método: Se estudió a 127 individuos control y 283 pacientes con trastornos neurológicos de origen desconocido. El análisis de NT se realizó mediante HPLC con detección electroquímica y el análisis de pterinas y 5-MTHF, mediante HPLC con detección de fluorescencia. Resultados: Se ha diagnosticado 3 deficiencias de tirosina hidroxilasa en una misma familia, 2 casos de distonía sensible a L-dopa, 2 familias con defiencia de guanosinatrifosfato-ciclohidrolasa dominante (14 casos), 2 deficiencias del transportador de glucosa y 43 deficiencias de folato en LCR. Conclusiones: Este estudio ha permitido el diagnóstico de nuevos pacientes y, lo que es más importante, el establecimiento en todos ellos de un tratamiento farmacológico o nutricional. Las deficiencias de 5-MTHF han sido las más frecuentes y se las ha detectado en diferentes grupos de pacientes.
Palabras clave:
Neurotransmisores
Dopamina
Serotonina
Pterinas
Líquido cefalorraquídeo
Transporte de glucosa
Transporte de folato
Barrera hematoencefálica
Background and objective: In the last few years, it has been described inborn errors of neurotransmitter and pterin metabolism and defects in folate and glucose transport across blood brain barrier. All these defects are classified as rare diseases and needs cerebrospinal fluid (CSF) sample analysis for diagnosis. Our aim was to evaluate the results of the application of a CSF analysis protocol in a pediatric population from Spain and Portugal presenting with neurological disorders of unknown origin. Patients and method: We studied CSF samples from and 283 patients with neurological disorders of unknown origin and 127 controls. Neurotransmitters were analysed by HPLC with electrochemical detection, and pterins and 5-methyltetrahydrofolate were determined by HPLC with fluorescence detection. Results: We diagnosed 3 patients with tyrosine hidroxylase deficiency, 2 with dopa responsive dystonia, 14 with GTP-ciclohydrolase deficiency, 2 with glucose transport deficiency and 43 with cerebral folate deficiency. Conclusions: This study allowed us to diagnose new patients, and more importantly, the establishment in all of them of a pharmacological or nutritional treatment. The most frequent defect found was CSF 5-methyltetrahydrofolate deficiency, which was present in different groups of patients.
Keywords:
Neurotransmitters
Dopamin
Serotonine
Pterins
Cerebrospinal fluid
Glucose transport
Folate transport
Blood brain barrier
Artículo
Opciones para acceder a los textos completos de la publicación Medicina Clínica
Suscriptor
Suscribirse

Comprar
Comprar acceso al artículo
Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora
Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora
Contactar
Teléfono para suscripciones e incidencias
De lunes a viernes de 9h a 18h (GMT+1) excepto los meses de julio y agosto que será de 9 a 15h
Llamadas desde España
932 415 960
Llamadas desde fuera de España
+34 932 415 960
E-mail
