




The objective of this project was to adapt to our setting following a systematic process based on the ADAPTE method the first clinical practice guidelines on X-linked hypophosphatemia (XLH) that were published in 2019.
Materials and methodsThe adaptation of the guidelines to our application and implementation setting was carried out in three phases —start-up, adaptation, and finalization— by a group of experts involved in the management of patients with XLH.
ResultsFollowing the original guide, the recommendations agreed by the group that elaborated the guidelines for diagnosis, frequency and scope of visits and specific follow-up in children and adults are presented. On the other hand, recommendations are established for both age groups with conventional treatment, as well as with burosumab in children or adults and those related to the controversial use of growth hormone in children. Suggestions are also proposed regarding the monitoring and management of musculoskeletal disorders and orthopedic treatment in children, dental health and hearing, and neurosurgical complications. Finally, a series of questions and areas are raised in order to deepen the possible future investigation.
ConclusionsThese recommendations constitute the systematic adaptation to our setting of the first evidence-based clinical practice guide for the diagnosis and management of XLH and we hope that they can contribute to the adequate management of the disease.
El objetivo de este proyecto fue adaptar la primera guía de práctica clínica en la hipofosfatemia ligada al cromosoma X (XLH) aparecida en 2019 a nuestro medio siguiendo un proceso sistemático basado en el método ADAPTE.
Materiales y métodosLa adaptación de las guías a nuestro ámbito de aplicación e implementación se llevó a cabo en 3 fases —puesta en marcha, adaptación y finalización— mediante un grupo de expertos implicados en el manejo de los pacientes con XLH.
ResultadosSiguiendo la guía original, se presentan las recomendaciones acordadas por el grupo elaborador de las guías para el diagnóstico, la frecuencia y el ámbito de las visitas y el seguimiento específico en niños y adultos. Por otro lado, se establecen las recomendaciones para ambas franjas de edad con tratamiento convencional, así como con burosumab en niños o adultos y las relacionadas al controvertido uso de hormona de crecimiento en niños. También se proponen sugerencias en cuanto al seguimiento y al manejo de las alteraciones del aparato locomotor y tratamiento ortopédico en niños, la salud dental y la audición y las complicaciones neuroquirúrgicas. Por último, se plantean una serie de cuestiones y áreas en las que profundizar en una posible investigación futura.
ConclusionesEstas recomendaciones constituyen la adaptación sistemática a nuestro medio de la primera guía de práctica clínica basada en la evidencia para el diagnóstico y el manejo de la XLH, y esperamos que pueda contribuir al manejo adecuado de la enfermedad.
X Chromosome-linked hypophosphatemia (XLH) or hypophosphatemic rickets is a dominant hereditary disease of phosphate metabolism characterized by a mutation in the phosphate regulating endopeptidase homolog X-linked (PHEX) gene, which is predominantly expressed in osteoblasts, osteocytes and odontoblasts1. Although rare, there are cases of XLH without a family history that are caused by a de novo mutation of the gene responsible for the disease1. Mutations in the PHEX gene lead to an increase in the concentration of fibroblast growth factor 23 (FGF-23), which regulates phosphate reabsorption in the kidneys and is considered potentially involved in many of the manifestations of XLH2.
XLH has an incidence of 3.9 cases per 100,000 live births and a prevalence that varies between 1.7 per 100,000 children and 4.8 per 100,000 in children and adults3. A recent study conducted in the United Kingdom shows that, in that country, between 1995 and 2016, the prevalence has been increasing, possibly as a consequence of an improvement in clinical practice and diagnosis4. The clinical manifestations of the disease vary greatly, from cases where there is nothing more than hypophosphatemia without associated bone symptoms, to cases with severe symptoms1. The most common clinical manifestations are growth retardation and gait disturbances, the so-called waddling gait, knee deformities such as genu varum, rickets, bone pain, abnormal dental development and occurrence of spontaneous dental abscesses, hearing loss, enthesopathy and osteoarthritis associated with lower limb and spinal deformities1,5,6. Diagnosis is made based on physical examination, laboratory tests, imaging tests, and family history; genetic analysis can confirm the diagnosis if the mutation is identified, but is not strictly necessary for diagnosis1.
Conventional, non-curative treatment consists of oral administration of phosphate in combination with calcitriol or alfacalcidol, in many cases requiring corrective surgery for bone deformities, dental treatment or even the administration of growth hormone7,8. The disease is associated with a significant deterioration in the quality of life in both children and adults, in which structural abnormalities play a key role9, associated with pain and functional limitation that persist despite treatment10. Although studies in this regard are scarce, patients with XLH seem to show an increase in mortality11. Very recently, burosumab, a human monoclonal antibody targeting FGF-23, has been added to the therapeutic arsenal for the management of this disease, showing efficacy and improving the symptoms of rickets as assessed by the rickets severity score (RSS) or the global impression of radiographic change, growth and biochemical abnormalities with respect to conventional treatment12–17.
For quite some time, both clinicians and patients have expressed concern about the lack of information on this disease, and the absence of clinical practice guidelines, a situation that has led to problems in its diagnosis and treatment18. Despite individual attempts by some authors to systematize the management of the disease8.18, it was not until 2019 that the first evidence-based clinical practice guideline for the diagnosis and management of XLH was published3. There are no published recommendations on this issue in our country. For this reason, a committee of experts with experience in the diagnosis and treatment of this entity was established to adapt this first 2019 guidelines to our setting following a systematic process based on the ADAPTE method. The project has been endorsed by the Spanish Association of Pediatric Nephrology (AENP), by the Spanish Association for the Study of Congenital Metabolic Errors (AECOM), by the Spanish Society of Pediatric Endocrinology (SEEP) and by the Spanish Bone and Mineral Metabolism Research Society (SEIOMM).
MethodsThe ADAPTE method was proposed as an alternative to improve efficiency in the development of clinical practice guidelines, avoiding the investment of large resources in the development of new guidelines when there are already guidelines adequately developed in other settings by different organisations for the same health problem19.
The adaptation process follows a well-established systematic approach that aims to endorse existing recommendations from other guidelines or to modify them to suit a different scope of application and implementation than the original one in which the source guidelines were developed. The adaptation takes place in 3 phases19: 1) Set-up phase, which consists of identifying the skills and resources needed to carry out the process, in order to determine whether adaptation is feasible; 2) adaptation phase, in which specific issues are identified, guidelines are searched and retrieved, the quality of the guidelines are assessed, their up-to-dateness, content and applicability are evaluated, decisions on adaptation are made and a draft adapted guideline is prepared; and 3) finalisation phase, in which feedback can be obtained from decision-makers who will be impacted by the guidelines, a process for reviewing and updating the adapted guidelines is established, and the final document is created. The full procedure for the development of guidelines following the ADAPTE process can be found elsewhere19.
Set-up phaseThe project was carried out by a group of experts involved in the management of XLH patients and included an expert in paediatric endocrinology (SMB), an endocrinologist with expertise in bone metabolism (GM), 2 experts in paediatric nephrology (DG-L and MIY), a rheumatologist with expertise in metabolic bone disease (PP) and an expert in paediatric dentistry (AL). These professionals constituted the guidelines development group (GDG) and in a first meeting they established the work plan for the development of the guidelines with the help of an external collaborator with expertise in research methodology (FR-V, see acknowledgements).
Adaptation phaseSearch, screening and evaluation of guidelinesGiven the existence of only one evidence-based guideline, the “Clinical practice recommendations for the diagnosis and management of X- linked hypophosphatemia”, this phase was limited to an initial assessment of the interest of these guidelines for professionals in our setting which was discussed at the first meeting mentioned above.
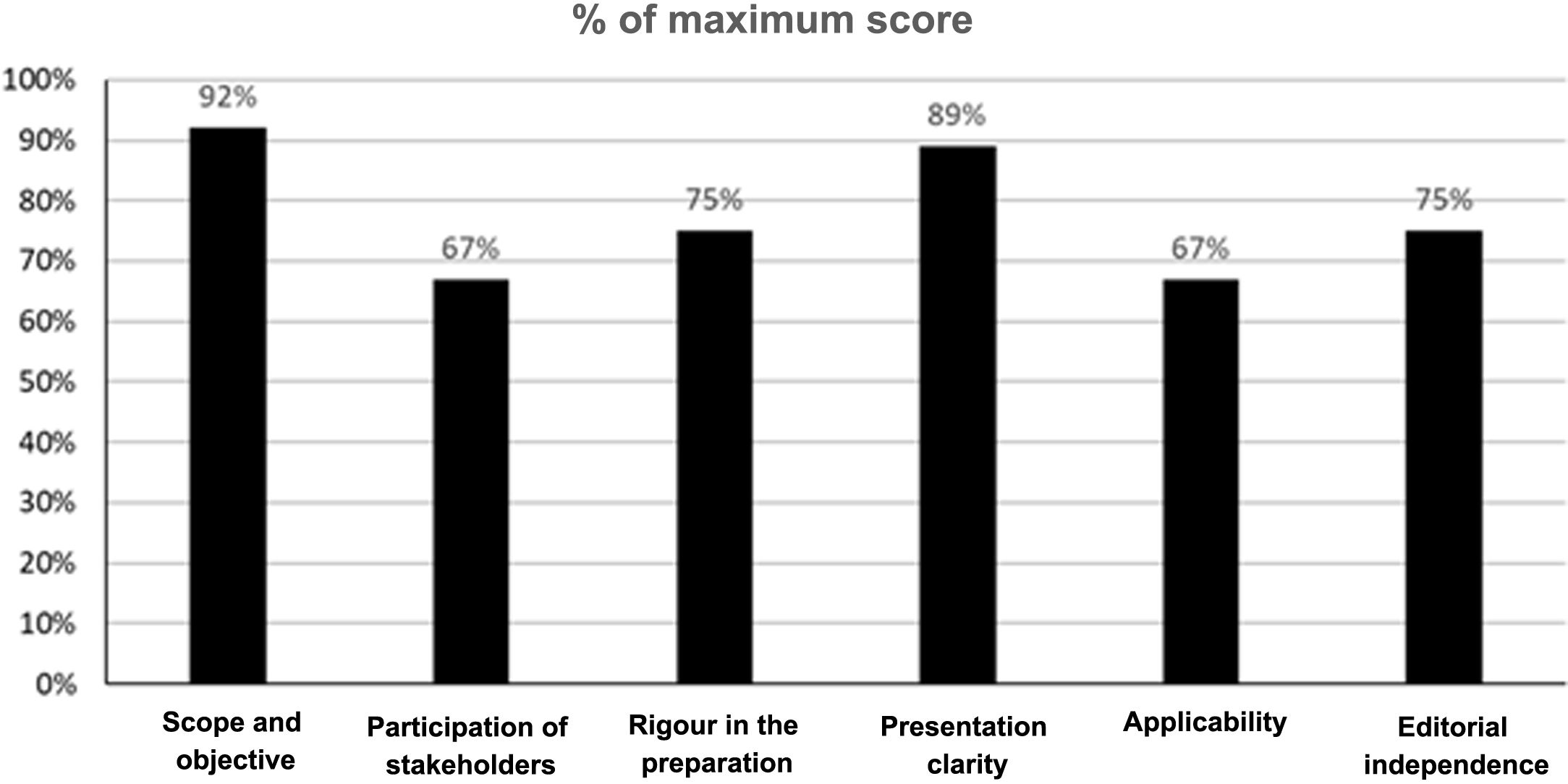
In addition, 2 members of the GDG carried out an evaluation of the quality of these guidelines using the tool Appraisal of Guidelines for Research and Evaluation II (AGREE II)20. The AGREE II is a tool that assesses the development and communication process of the guidelines. It consists of 23 items assessing the degree of agreement on a 7-point Likert scale, from one strongly disagree to 7 strongly agree. The instrument evaluates 6 domains: scope and purpose, stakeholder involvement, rigour of development, clarity of presentation, applicability and editorial independence. In addition, an overall assessment of the quality of the guideline is included which is also answered on a 7-point Likert scale (from one “lowest possible quality” to 7 “highest possible quality”), and the evaluators have to answer the following question “Would you recommend this guideline for use?” with 3 alternative answers (i.e. yes, yes with modifications or no). For the pre-assessment training, the assessors used the training videos and the explanatory manual of the instrument available in the AGREE II (Available at: https://www.agreetrust.org/). The determination of the aggregate score of the 2 assessors was carried out according to the instructions accompanying the instrument. The overall results of the evaluation of the guidelines are presented in Fig. 1. Inter-rater agreement was not high, with an intra-class correlation coefficient of 0.42 (Appendix Supplementary Table 1). However, both evaluators agreed in assessing the overall quality of the guidelines with a score of 6 on a 7-point Likert scale, and both agreed in recommending the guidelines for use.
Selection and preparation of recommendations
In order to have up-to-date information on the management of XLH, an update of the literature search of the original guidelines was carried out. For this, like the original guidelines, a PubMed search was conducted from 26 June 2018 to 24 July 2019 using a search strategy somewhat broader in its terms than that of the original article (Appendix B Supplementary Table 1). The new search yielded 82 records that were distributed among all the members of the GDG.
All recommendations from the original guidelines were compiled in English in a table following the same subject structure as the guidelines. This table was distributed to all GDG members so that they could review the recommendations and individually indicate whether they agreed with them in their current wording, whether they required any modification and, if so, the reason for such modification; if they considered that due to their experience or specialty they could not assess a certain recommendation, they marked it as not evaluable. The individual evaluations of all evaluators were compiled in a single table which was reviewed and discussed by all evaluators in 2 teleconferences to reach an overall agreement of the GDG on each of the recommendations where any of the evaluators had indicated that some modification was required or had included a comment on a recommendation; recommendations where there was a consensus of all evaluators to accept them without modification were not discussed and were considered accepted. Once the modifications or comments to be added to those recommendations that required it were agreed, the recommendations were translated into Spanish. This translation was reviewed by all GDG members and discussed in a new teleconference. Ultimately, a final document was produced that included the original recommendation in English, the Spanish translation, whether or not it required modification, the modified recommendation and the rationale for the modification (Appendix B Supplementary Table 2).
Completion phaseFollowing publication of the guidelines, any reader interested in the subject can send their comments to the author for correspondence to be taken into account in future updates of the guidelines.
Levels of evidenceThe levels of evidence included with the recommendations are those reported in the original guidelines and are based on those used by the American Academy of Pediatrics in their guidelines21 (Appendix B Supplementary Table 2).
ResultsRecommendations for diagnosis (Table 1)According to the guidelines, diagnosis is established by clinical, radiological and biochemical assessment, although confirmation by genetic testing of the PHEX gene in children and adults is recommended if feasible. The determination of FGF-23 can be useful to support the diagnosis, but it must be taken into account that normal levels do not exclude the diagnosis23, and that conventional treatment with phosphate and vitamin D can alter FGF-23 levels23,24.
Recommendations for diagnosis.
| - In children, a diagnosis of XLH should be considered in the presence of clinical and/or radiological signs of rickets, growth failure, and serum phosphate levels below age-adjusted reference values, associated with renal phosphate wasting and no vitamin D or calcium deficiency (grade B, moderate recommendation) |
| - In adults, the diagnosis of XLH should be considered in the presence or with a history of lower limb deformities, and/or clinical and/or radiological signs of osteomalacia (including pseudofractures, early osteoarthritis and enthesopathy) in the context of serum phosphate levels below age-adjusted reference values associated with renal phosphate wasting (grade B, moderate recommendation) |
| - We recommend that any first-degree family member of a patient with XLH be screened for XLH (grade D, weak recommendation); male children of XLH-affected men are not at risk |
| - We recommend carrying out the following initial diagnostic procedures (grade B, moderate recommendation): |
| • A detailed clinical evaluation, including evidence of rickets, growth retardation, dental abnormalities, and signs of craniosynostosis and/or intracranial hypertension using fundus imaginga |
| • A radiological evaluation to diagnose and assess the degree of rickets (rickets severity score [RSS]) and osteomalacia lesionsa |
| • Biochemical tests including serum levels of phosphate, calcium, alkaline phosphatase, parathyroid hormone, 25(OH) vitamin D, 1,25(OH)2 vitamin D, intact FGF-23, and creatinine, as well as urinary levels of calcium, phosphate, and creatinine from a spot urine sample for the determination of tubular reabsorption of phosphate (TRP)band urinary calcium:creatinine and phosphate:creatinine ratiosa |
| - We recommend that non-selective renal tubular phosphate wasting (suggesting renal Fanconi syndrome) be ruled out by looking for abnormal bicarbonate, amino acid, glucose and/or uric acid losses in urine samples and low molecular weight proteinuria (grade B, moderate recommendation) |
| - We recommend confirming the clinical diagnosis of XLH by genetic analysis of the PHEX gene in children and adults if possible (grade B, moderate recommendation) |
| - If genetic analysis is not available, elevated plasma levels of intact FGF-23 and/or a positive family history of XLH support the diagnosis (grade C, moderate recommendation) |
| - We recommend considering other causes of hereditary or acquired hypophosphatemia if the PHEX gene test yields a negative result for XLH (grade B, moderate recommendation) |
| - We recommend that genetic counselling be offered to patients with XLH, especially in the transition from childhood to adult care, as well as to families planning a pregnancy (grade C, moderate recommendation) |
| - Methods to detect PHEX mutations can be applied to preimplantation genetic diagnosis or prenatal diagnosis. However, recommendations should be tailored to country-specific legal and ethical standards and communicated using appropriate genetic counselling (grade D, weak recommendation) |
| - We recommend further testing after diagnosis, including studies aimed at diagnosing the presence and severity of common and rare complications summarized in **** (grade C, moderate recommendation) |
| - Before transition to adult care, we suggest performing an echocardiogram to assess cardiovascular statusa |
Recommendations that have undergone some modification from the original. Where the modification is a change to the text, it is underlined; deletion of the text is not reflected in the table and is discussed in the explanatory text.
Tubular reabsorption of phosphate is calculated as TRP = 1-[PO4 (U) × Cr (S)]/[PO4 (S) × Cr(U)]; where Cr, creatinine; S, serum; U, urine; PO4, phosphate1,22.
Among the biochemical tests Table 2 we have included the phosphate:creatinine ratio in spot urine. The 24 h urinary phosphate excretion (24 h UPE) is considered the standard for assessing phosphate intake. In young children, the phosphate:creatinine ratio in spot urine is potentially interchangeable with the 24 h urine sample25. And any urine index obtained from a spot urine test is of greater clinical value in young children than a 24 h urine test26.
Initial evaluation of common and rare complications of XLH.
| Clinical evaluations |
| - Growth chart |
| - Signs of rickets and/or deformity in the legs |
| - Measure intermalleolar and intercondylar distance |
| - Head circumference and skull shapeb |
| - Neurological examination (for consequences of craniosynostosis and spinal stenosis) |
| - Hearing evaluationa |
| - Oral examination |
| - Musculoskeletal function (gait)a,b |
| Biochemical evaluations |
| - Blood: calcium, phosphate and creatinine |
| - Urine (spot sample): calcium, phosphate and creatinine |
| - TmP/GFR |
| - Estimated glomerular filtration rate |
| - 25(OH) vitamin D |
| - 1,25(OH)2 vitamin D |
| -PTH |
| - Alkaline phosphatase (children and adults) and bone alkaline phosphatase (adults) |
| - Intact FGF-23 (in case of negative family history) |
| Imaging Assessments |
| - Wrist and/or knee and/or ankle X-rays (rickets)b |
| - Standardised and well-positioned anteroposterior X-ray of standing limb alignment (using low-dose techniques, if possible). |
| - Panoramic radiographa |
| - Brain MRI |
| - Kidney ultrasound (nephrocalcinosis) |
FGF-23: fibroblast growth factor 23; PTH: parathyroid hormone; TmP/GFR: maximal tubular phosphate reabsorption per glomerular filtration rate; XLH: X-linked hypophosphatemia.
All assessments are applicable to children aged < 5, children 5–18 and adults except where otherwise specified.
In addition to the evaluations previously recommended by the original guidelines, the GDG suggests performing an echocardiogram at the time of transition to adult care, given the possible cardiovascular abnormalities that these patients may have. Among the morbidities associated with XLH and those related in some way to FGF-23, the possible presence of cardiovascular calcification and hypertension has been reported2. In addition, some studies indicate that about 20% of subjects with XLH have echocardiographic results suggestive of left ventricular hypertrophy27. Other authors, in case series, have also found left ventricular hypertrophy and blood pressure abnormalities, and suggest that these patients should be closely monitored to assess the development of these two abnormalities28. As a general observation, it has also been pointed out that in addition to screening for renal, rheumatological, dental and neurosurgical disorders, screening for cardiovascular disorders should also be added29.
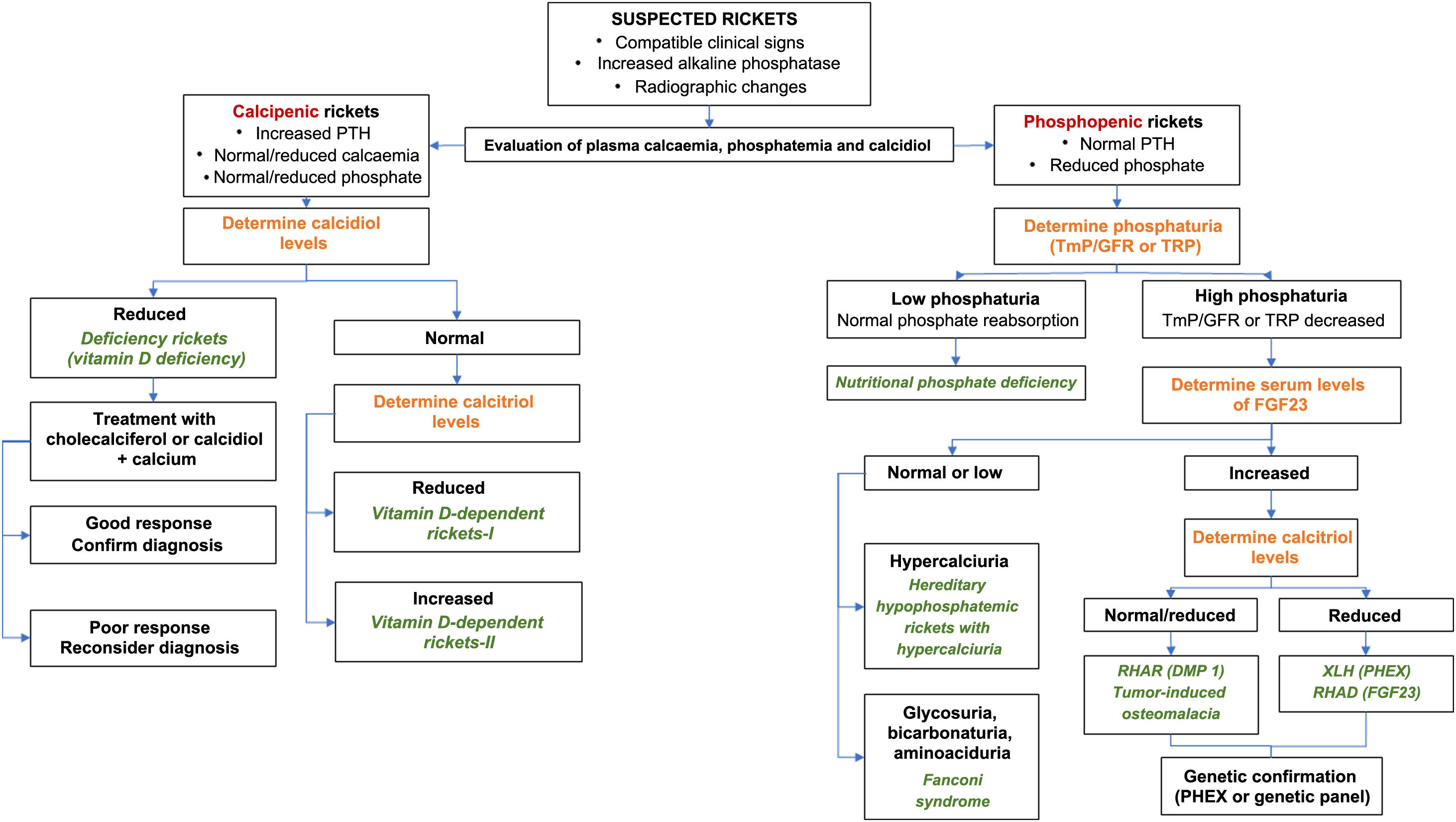
The algorithm in Fig. 2 summarizes the diagnostic management of XLH.

Diagnostic algorithm for rickets.
FGF-23: fibroblast growth factor 23; PTH: parathyroid hormone; RHAD: autosomal dominant hypophosphatemic rickets; ADHR: autosomal dominant hypophosphatemic rickets; GFR: glomerular filtration rate; TmP: maximal tubular phosphate reabsorption; XLH: X chromosome-linked hypophosphatemic rickets.
Source: Translated and adapted with permission of Springer Nature Customer Service Centre GmbH: Goodbye, Advances in Therapy. González-Lamuño D45. ©Springer Healthcare Ltd., part of Springer Nature 2020.
Below, general recommendations are made for the follow-up of both children and adult patients (Tables 3–5). However, it should be borne in mind that the manifestations and complications of the disease vary greatly from patient to patient, and follow-up should be individualised according to the patient's medical history, clinical manifestations, stage of development and medical judgement.
Frequency and scope of visits.
| - We suggest that multidisciplinary teams provide patient care (grade D, weak recommendation)a |
| - We suggest screening children with XLH at least every 3 months during phases of rapid growth (early childhood [0−2 years] and puberty [10−16 years]) or after initiation of treatment (grade C, weak recommendation)a |
| - We suggest that adult patients be screened every 6 months if they receive treatment, or once a year if they do not receive drug therapy (grade C, weak recommendation). |
Follow-up of children with XLH (grade C, moderate recommendation).
| - We recommend measuring height, weight, head circumference (up to 5 years of age), intercondylar and intermalleolar distances, and blood pressure |
| - We recommend calculating the body mass index (BMI) and the annual growth rate. |
| - We recommend documenting head shape and history of headaches, dental abscesses or maxillofacial cellulitis, bone pain, fatigue, and physical activity level |
| - We recommend that an orthopaedic assessment of the locomotor system should be performed in the presence of lower limb deformity (varus, valgus or anteroposterior) |
| - We recommend looking for evidence of hearing loss, spinal deformity and scoliosis, manifestations related to craniosynostosis, Chiari 1 malformation and/or intracranial hypertension and maxillary dysmorphism |
| - We recommend assessing bone age (preferably by radiography of the carpal bones and the Greulich and Pyle Atlas) to assess growth potential in children > 5 years with growth retardationa |
Follow-up of adults with XLH (grade C, moderate recommendation).
| - We recommend measuring height, weight and blood pressure and calculating BMI |
| - We recommend recording a history of headaches, oral manifestations (including periodontal disease, dental abscess or maxillofacial cellulitis), musculoskeletal pain, pseudofractures, fatigue and level of physical activity |
| - We recommend looking for evidence of hearing loss, enthesopathy, osteoarthritis, spinal deformity and scoliosis, muscle impairment, range of motion, manifestations related to Chiari 1 malformation and/or intracranial hypertension |
The original guidelines recommends that multidisciplinary teams be coordinated by an expert in metabolic bone diseases. The GDG considers that this recommendation, although appropriate, is not fully adapted to the reality of our context, where, depending on the area, such experts may not be available. We believe that each centre must decide who will be the coordinator of the multidisciplinary team.
In addition, the original guidelines suggest spacing out follow-up in those patients who have a positive response to treatment. The GDG considers that follow-up depends more on the patient's age or specific situations, such as being in the transition period to adult care, rather than on the treatment they receive.
Among the recommendations for monitoring children is the assessment of bone age. The GEG considers it important to emphasise that this assessment should be carried out by reliable methods and recommends radiography of the carpal bones and the use of the Greulich and Pyle Atlas. A recent systematic review of 17 studies has shown a good degree of agreement between chronological and bone age determined using the Greulich and Pyle Atlas30. Thus, the differences between both ages were generally less than one year in all age groups30.
Wherever possible, these recommended follow-up tests should be accompanied by a quantification of severity; for example, with the use of visual analogue scales to assess pain intensity or fatigue, or the 6 min walk test (6 MWT) to assess functional capacity and for which manuals are available for use in other chronic diseases31 that can be adapted to this setting, as there are no specific recommendations for XLH patients. However, there are reference values of 6 MWT for children and adolescents between 4–19 years32; below that age it is not feasible to carry it out.
Follow-up of all patients (children and adults) (Table 6)In burosumab-treated patients, we recommend monitoring fasting serum phosphate levels along with TRP and maximal tubular phosphate reabsorption for TmP/GFR. The reason for this recommendation is that in the work-up of hypophosphatemia it is recommended to first calculate the TRP, but in the case of normal-low TRP, the TmP/GFR is more accurate in detecting urinary phosphate loss33.
Follow-up of all patients (children and adults).
| - We recommend check-ups by the dentist twice a year after tooth eruption or from the first year of life to prevent and treat dental infections and periodontitis (grade C, moderate recommendation)a |
| - We recommend monitoring blood levels of alkaline phosphatase (AP) (serum levels of total AP in children and adults and, optionally, bone-specific AP in adults), calcium, phosphate, creatinine, PTH, and 25(OH) vitamin D (grade B, moderate recommendation)a. We recommend measuring urinary calcium and creatinine levels to estimate the calcium:creatinine ratio in patients receiving conventional therapy or burosumab (grade B, moderate recommendation) |
| - In patients treated with burosumab, we recommend monitoring fasting serum phosphate levels (grade B, moderate recommendation) along with TRP and maximal tubular reabsorption of phosphate per glomerular filtration rate (TmP/GFR) (grade B, weak recommendation) every 2 weeks for the first month of treatment, and every 4 weeks for the next 2 months and thereafter every 3−6 months; we also recommend measuring fasting serum phosphate level 4 weeks after dose adjustment (grade B, moderate recommendation) and suggest measuring serum 1,25(OH) levels2 vitamin D every 6 months analysed together with urinary calcium excretion as safety parameters (grade B, weak recommendation) |
| - We recommend assessment of disease severity by wrist and knee radiographs in children (using RSS) who do not respond well to treatment or whose bone deformities worsen despite drug therapy, in children who may need orthopaedic surgery, in those complaining of unexplained bone pain, or in adolescents with persistent lower limb deformities when transitioning to adult care. Radiographs should be standard anteroposterior views of the lower limbs in the upright position (using low-dose radiation when possible) to assess limb deformities, joint alignment, and bone quality (grade B, moderate recommendation)a |
| - In patients receiving conventional or burosumab treatment, we recommend renal ultrasound at least every 2 years in those without nephrocalcinosis, and annually in patients with nephrocalcinosis and/or persistent hypercalciuria (grade C, moderate recommendation)b |
| - We recommend a brain MRI (if possible, including a hypointense bone sequence ["black bone imaging sequence"] to capture images of the skull) if the morphology of the skull suggests craniosynostosis or clinical signs of intracranial hypertension (grade C, moderate recommendation). |
| - We recommend performing a panoramic radiograph (radiography of upper and lower jaws and teeth) at 5 years of age and in adults with recent oral manifestations. Radiographs should be repeated according to individual needs; retrocoronal and periapical radiographs and cone-beam computed tomography may be used to detect and monitor endodontic, periodontal, or peri-implant infections (grade D, weak recommendation) |
| - To assess bone health in patients with XLH, we do not recommend the routine use of dual-energy X-ray absorptiometry (DXA) or peripheral quantitative computed tomography (pQCT) (grade C, moderate recommendation) |
| - We suggest providing patients with contact details of patient associations, informing them of scientific developments (including new treatments), supporting educational and professional input and providing social support (grade D, weak recommendation). |
| - We suggest introducing the 6 MWT and assess quality of life with the age-appropriate version of the EuroQol-5D in patients from 5 years of age at yearly intervals or every 2 years whenever facilities are available (grade D, weak recommendation)to. |
Among the recommendations for all patients is the determination of urinary calcium and creatinine for the calculation of the calcium:creatinine ratio, which is important for the follow-up of patients receiving conventional treatments (for example, vitamin D) or burosumab. The GDG considers that this determination should preferably be made in 24 h urine, at least in adults, since in spot urine, the fasting calcium:creatinine ratio underestimates calciuria whereas it may overestimate it in postprandial urine. It can also be performed on two spot urine samples.
As has been commented for other evaluations, the GDG considers that whenever possible the use of specific evaluation instruments should be recommended, such as the RSS for the evaluation of severity, or the use of quality-of-life questionnaires such as the family of EQ-5D tools that have been developed for both adults (EQ-5D-5L) and children (EQ-5D-Y). The EQ-5D evaluates 5 domains (mobility, self-care, usual activities, pain/discomfort and anxiety/depression) at 5 levels (EQ-5D-5L) or at 3 levels (EQ-5D-Y)34. They are easy to use, reliable and sensitive to change, and are available in Spanish for free use as long as there is no commercial purpose34.
Therapeutic recommendations in children (Table 7)The GDG is in global agreement with the therapeutic management recommendations for children with XLH. However, it considers it important to make some modifications that are discussed below, especially with regard to the dosage of some interventions:
- -
We believe that the frequency of phosphate supplementation should not be determined solely on the basis of AP, since it is not the most important factor for monitoring the effects of treatment; AP may not have returned to normal and a reduction in phosphate supplementation may be necessary35.
- -
The recommendation to use low doses of phosphate seems appropriate, but we believe it is important to emphasise that these should be therapeutic doses to avoid under-dosing.
- -
Although not specified in the recommendations, urinary calcium excess can be avoided by adjusting the dose of vitamin D and phosphate and introducing thiazides if necessary36,37.
- -
We believe that the dose of active vitamin D should always be established based on the weight of the patient
Therapeutic recommendations in children.
| - We recommend treating children who have an overt XLH phenotype with a combination of oral phosphorus (phosphate salts) and active vitamin D (calcitriol or alfacalcidol) as soon as the diagnosis is made (grade B, moderate recommendation) |
| - We recommend an initial dose of 20−60 mg/kg of body weight per day (0.7−2.0 mmol/kg per day) of elemental phosphorus in infants and preschool children, which should be adjusted as rickets, growth, and AP and parathyroid hormone (PTH) levels improve (grade C, moderate recommendation) |
| - We recommend administering phosphate supplementation as often as possible, for example, 4−6 times a day in young patients with high levels of AP (grade B, moderate recommendation)a |
| - We recommend a progressive increase in the dose of phosphate supplementation in case of insufficient clinical response, although doses higher than 80 mg/kg per day (depending on elemental phosphorus) should be avoided to prevent gastrointestinal discomfort and hyperparathyroidism. If these side effects occur, treatment should be adjusted by lowering the dose and/or increasing the frequency (grade C, moderate recommendation) |
| - We recommend the use of low doses within the effective dose range (that is, 20−60 mg/kg) in patients with mild phenotypes, for example, infants diagnosed by familial screening (grade C, moderate recommendation)a |
| - We recommend an initial dose of calcitriol of 20−30 ng/kg of body weight daily or alfacalcidol 30–50 ng/kg of body weight daily (grade C, moderate recommendation)a |
| - To prevent nephrocalcinosis, we recommend keeping calciuria levels within the normal range (avoid exceeding more than 4 mg/kg body weight in 24-h urine in children older than 2 years who are continent) and avoid high doses of phosphate supplements; we suggest measures to decrease urinary calcium concentration, excretion, and/or crystallization if necessary, including regular water intake, administration of potassium citrate, and limited sodium intake (grade C, moderate recommendation)a |
| - Regarding secondary hyperparathyroidism, we recommend the following: |
| • Patients receiving conventional treatment with elevated PTH levels will have the dose of active vitamin D increased and/or the dose of oral phosphate supplements decreased (grade C, moderate recommendation) |
| • Calcimimetic treatment could be considered in patients with persistent secondary hyperparathyroidism despite the above measures (grade D, weak recommendation)a,b. Cinacalcet should be used with caution in XLH, as it has been associated with serious adverse reactions, namely hypocalcaemia and QT interval increase (grade X, strong recommendation) |
| • Parathyroidectomy should be considered in tertiary hyperparathyroidism (persistent hypercalcaemic hyperparathyroidism) despite optimized treatment with active vitamin D and cinacalcet (grade C, moderate recommendation) |
| - We suggest that patients with vitamin D deficiency receive natural vitamin D supplements (cholecalciferol) (grade C, weak recommendation)a |
| - We do not recommend routine calcium supplementation in children with XLH, although a dietary assessment of daily calcium intake should be performed (grade D, weak recommendation) |
| - We recommend that treatment planning be carried out in a multidisciplinary team setting prior to surgery; we also suggest reducing or discontinuing active vitamin D supplementation if patients are immobilised for a long period and restarting it as soon as the patient resumes mobility (grade D, weak recommendation)to |
In addition, for both recommendations in children and adults, ergocalciferol has been eliminated as natural vitamin D supplements, since it is not routinely used in our setting.
Finally, for the management of secondary hyperparathyroidism, given that the original guidelines suggest a possible use of Cinacalcet, it has been specified that this use is not included among the product's indications; in our setting, this drug is indicated for the treatment of secondary hyperparathyroidism (HPT) in children 3 years of age or older, with chronic renal failure on dialysis in whom secondary HPT is not adequately controlled with the usual treatment.
Recommendations for the administration of burosumab in children with XLH (Table 8)Following the recent final decision of the European Commission (on 30 September 2020), burosumab has obtained authorisation for the extension of its indication to the treatment of XLH in children and adolescents aged 1–17 years with radiographic signs of bone disease, and in adults38. The recommendations of the original guidance are aligned with this indication in the present document, following the also recent agreement for national reimbursement at the time of submission for publication. The recommended starting dose of burosumab has been changed from 0.4 to 0.8 mg/kg/every 2 weeks for the paediatric population based on the results of the latest clinical trials with this drug12,16. However, when burosumab is prescribed, the recommendations included in the SmPC must be taken into account39.
Recommendations for the administration of burosumab in children with XLH.
| - If available, we recommend considering burosumab treatment in children with XLH ≥ 1 year and in adolescents in the following situations: radiographic evidence of overt bone disease and disease refractory to conventional therapy; or complications related to conventional therapy; or the inability of the patient to adhere to conventional therapy, assuming adequate follow-up is feasible (grade B, moderate recommendation)a |
| - In children, we recommend a starting dose of burosumab of 0.8 mg/kg of body weight (rounded to the nearest 10 mg), administered subcutaneously every 2 weeks (grade B, moderate recommendation)a |
| - We recommend gradually increasing the dose of burosumab in increments of 0.4 mg/kg to raise fasting serum phosphate levels around the lower limit of the age-adjusted normal reference range up to a maximum dose of 2.0 mg/kg of body weight (maximum dose 90 mg) (grade B, moderate recommendation) |
| - Burosumab should not be adjusted more often than every 4 weeks (grade B, moderate recommendation). |
| - During the dose titration period, we suggest monitoring fasting serum phosphate levels between injections, ideally 7−11 days after the last injection, to detect the presence of hyperphosphatemia; after reaching steady state, which can be assumed to be after 3 months of receiving a stable dose, fasting serum phosphate levels should be assessed preferably just before injections to detect underdosing (grade B, weak recommendation) |
| - The dose should be discontinued if the fasting serum phosphate level is above the upper limit of the normal range. Burosumab can be restarted at half the previous dose when serum phosphate concentration is below the normal range (grade B, moderate recommendation) |
| - We recommend that burosumab not be co-administered with conventional therapy when fasting phosphate levels prior to initiation of therapy are within the age-adjusted normal reference range, or in the presence of severe renal failure (grade X, moderate recommendation) |
Growth hormone has been used in these patients as adjuvant therapy. However, the response to treatment has been variable and it has not been possible to adequately demonstrate that the benefits outweigh the risks, hence the limitation of its use to very restricted populations18.
Growth hormone-related recommendations.
| - We do not recommend the routine administration of recombinant human growth hormone (rhGH) in patients with XLH (grade C, weak recommendation) |
| - Treatment with rhGH could be considered in children with short stature, provided that the levels of AP and parathyroid hormone were well controlled (grade C, weak recommendation) |
The GDG agrees with the recommendations of the guidelines in this regard. However, it does not consider that it is strictly necessary to recommend a reduction in the dose of vitamin D in all patients in whom long-term immobilization is anticipated, but rather that this decision should be made according to the clinical situation of each patient and, therefore, is included as a suggestion.
Recommendations for conventional treatment in adults.
| - We recommend that adults with XLH who are symptomatic receive treatment with active vitamin D together with oral phosphorus (phosphate salts) to reduce osteomalacia and its consequences and improve oral health (grade B, moderate recommendation). |
| - We suggest treating pregnant and breastfeeding women with active vitamin D in combination with phosphate supplementation if necessary (grade D, weak recommendation). |
| - We do not recommend routine treatment of adults with XLH who are asymptomatic (grade C, moderate recommendation) |
| - We recommend the use of substantially lower doses of active vitamin D and oral phosphate than those used in children (grade C, moderate recommendation). We recommend a dose range of 750−1,600 mg daily (based on elemental phosphorus) for phosphate and 0.50−0.75 and 0.75−1.5 µg daily for calcitriol and alfacalcidol, respectively (grade C, weak recommendation) |
| - We suggest reducing the dose of active vitamin D in patients with anticipated long-term immobilisation, to prevent hypercalciuria and hypercalcaemia (grade D, weak recommendation)to |
| - We recommend discontinuing phosphate supplementation in patients with significantly increased PTH levels (grade C, moderate recommendation) |
| - We suggest that active vitamin D could be administered to adult patients with secondary hyperparathyroidism without phosphate supplementation if carefully monitored (grade D, weak recommendation). |
| - We suggest administering natural vitamin D supplements (cholecalciferol) in case of vitamin D deficiency; we also suggest ensuring a normal calcium intake (grade C, weak recommendation)a |
As previously mentioned, burosumab is currently indicated for the treatment of XLH in children and adolescents aged 1–17 years with radiographic signs of bone disease, and in adults38. We consider it important to rule out that the pain is not caused by a degenerative condition unrelated to XLH when used in adults due to persistent bone and/or joint pain, as this does not respond to burosumab40. The management of this symptomatology resulting from degenerative changes is addressed in the following section.
Recommendations for burosumab treatment in adults.
| - If available for this indication, we recommend considering burosumab treatment in adults with XLH who have the following characteristics: persistent bone and/or joint pain due to XLH and/or osteomalacia limiting daily activities (ruling out that the pain is associated with other common conditions such as osteoarthritis, spondylarthrosis or canal stenosis, among others); pseudofractures or fractures related to osteomalacia; and insufficient or refractory response to conventional therapy (grade B, moderate recommendation)a |
| - We also recommend considering burosumab treatment if patients experience conventional treatment-related complications (grade D, weak recommendation) |
| - We recommend an initial dose of burosumab of 1.0 mg/kg of body weight (maximum dose of 90 mg) administered subcutaneously every 4 weeks (grade B, moderate recommendation) |
| - We suggest initial monitoring of fasting serum phosphate levels between injections, ideally 7−11 days after the last injection to detect the presence of hyperphosphatemia; after reaching a steady state, which is assumed to be achieved after 3 months of receiving a stable dose, serum phosphate levels should be assessed during the last week before the next injection to detect underdosing (grade B, weak recommendation) |
| - The dose should be discontinued if the fasting serum phosphate level is above the upper limit of the normal range. Burosumab can be restarted at half the previous dose when the serum phosphate concentration is below the normal range (grade C, moderate recommendation) |
| - Burosumab should not be administered together with conventional treatment in patients whose phosphate levels before the start of treatment are within the age-adjusted normal reference interval or in the presence of severe renal failure (grade X, moderate recommendation) |
Recommendations for the treatment of musculoskeletal system disorders.
| - We recommend interventions aimed at reducing bone and joint pain, deformity, stiffness, muscle weakness and improving walking distance and physical activity. These interventions include general measures such as the use of analgesics (e.g., the use of nonsteroidal anti-inflammatory drugs [NSAIDs] for short periods of time), intra-articular injections (in the presence of degenerative changes), physical therapy, rehabilitation, physical activity and non-pharmacological treatment of pain and the assessment of orthopaedic surgery according to progression and functional limitation (grade D, weak recommendation)a |
Recommendations for orthopaedic treatment in XLH children.
| - We do not recommend the use of splints or insoles for the treatment of lower limb deformity in children with XLH (grade C, moderate recommendation) |
| - We suggest insisting on the importance of resistance exercise supervised by qualified personnel, maintenance of joint range, and maximizing strength and endurance (grade D, weak recommendation)a |
| - We recommend physical therapy after surgery or in case of decreased range of motion, muscle weakness, fatigue, instability or if there is physical deconditioning (grade D, weak recommendation) |
| - We recommend that elective surgical treatment be performed only in children in whom medical treatment has been maximized for at least 12 months (grade C, moderate recommendation) |
| - We suggest that the surgery be performed by a surgeon with experience in metabolic bone diseases (grade B, moderate recommendation) |
| - Surgical treatment should be considered in the event of a persistent deformity (Zone 2 mechanical axis deviation or higher) despite optimized drug therapy and/or the presence of functional interference symptoms (grade C, weak recommendation) |
| - We recommend that the age of the child be considered as an important factor in the decision-making process: guided growth techniques depend on the remaining growth potential of the child and therefore should be carried out at least 2−3 years before skeletal maturity (14 years in girls and 16 years in boys), whereas complications associated with osteotomy are reduced when surgery is performed in late childhood or after skeletal maturity (grade C, moderate recommendation) |
| - We recommend that emergency surgical treatment, such as fracture fixation, be carried out when necessary (grade B, moderate recommendation) |
| - We suggest that, after surgery, regular clinical and functional evaluations, including radiography, be done at 12 months after surgery, or sooner if the bone deformity worsens and/or there is clinical concern. Additional evaluations should be performed intermittently until skeletal maturity (grade C, moderate recommendation) |
Recommendations for dental health management.
| - In children and adults with ongoing oral manifestations, we recommend treatment with active vitamin D and phosphate supplementation to improve dentin mineralization and reduce the number of dental abscesses and the severity of periodontitis (grade B, moderate recommendation) |
| - In children, in addition to standard prophylactic care, we recommend dental check-ups every 6 months and suggest sealing pits and fissures with liquid resin composite in primary and permanent teeth as early and as often as necessary (grade C, weak recommendation). If the paediatric patient has already had an abscess, prophylactic pulpotomy and crowns are recommendeda |
| - We suggest a thorough clinical work-up looking for pulpal necrosis (discoloration, fistula, swelling, abscess, cellulitis or pain) and performing retrocoronal and/or periapical radiographs or panoramic radiographs to look for enlarged pulp chambers and periapical bone loss depending on the findings of a clinical examination (grade B, weak recommendation) |
| - We suggest optimizing the conventional medical treatment of XLH before starting orthodontic treatment (grade C, moderate recommendation) |
| - In adults, we recommend visits twice a year for supportive conventional periodontal therapy, which should include periodontal risk assessment and supragingival and subgingival debridement if necessary (grade B, moderate recommendation) |
| - In adults, we suggest that dental implant surgery be carried out after 3−6 months of medical treatment, which should continue for 6 months after implant surgery; healing time should be extended to 6 months (grade D, weak recommendation) |
Recommendations for the management of hearing.
| - We suggest informing patients and families that hearing problems may occur, and that any suspected hearing loss should be carefully evaluated (grade D, weak recommendation) |
| - We suggest treating hearing loss in a similar way to other causes of peripheral hearing loss, with hearing aids, prevention of exposure to noise and avoidance of ototoxic medications (grade D, weak recommendation) |
Recommendations for the management of neurosurgical complications.
| - We suggest an annual basic neurological evaluation, but we do not recommend further examinations in patients with XLH who are asymptomatic (grade C, weak recommendation) |
| - We suggest that patients and families be informed that neurosurgical complications may occur and that any concern about central nervous system function should be reported and treated early (grade C, weak recommendation) |
| - We recommend a full evaluation with ophthalmoscopy and brain or cranial imaging in any patient with XLH who presents with cranial morphology suggestive of craniosynostosis or clinical symptoms of intracranial hypertension, lower brainstem compression or upper cervical cord compression (suggestive of Chiari 1 malformation) (grade C, moderate recommendation). |
Recommendations related to lifestyle.
| - We recommend that physical activity for XLH patients be encouraged and adapted to the patient's ability. All sports are allowed unless there are individual contraindications; aerobic activities are preferred because anaerobic activities can cause too much stress on the skeleton (grade D, weak recommendation) |
| - We support the same guidelines for the prevention and treatment of obesity that are given to the general population (grade D, weak recommendation) |
Topics for future research.
| - Develop a comprehensive registry of children and adults with XLH to assess the natural history of the disease, including rare complications and mortalitya |
| - Evaluate the impact of XLH on schooling, social life and professional activity |
| - Develop clinical and/or biological and/or radiological indices to support the evaluation of the efficacy and safety of the treatment |
| - Define the degree of skeletal deformity that is compatible with a good quality of life |
| - Define the risk-benefit ratio of surgical interventions (osteotomy versus guided techniques) |
| - Assess the risk-benefit ratio of conventional treatment in adults before and after surgical interventions |
| - Assess the risk-benefit ratio of conventional treatment in pregnant or breastfeeding women with XLH |
| - Assess the optimal target range of surrogate biochemical variables (such as fasting serum phosphate levels and maximal tubular reabsorption of phosphate per glomerular filtration rate) in patients receiving burosumab |
| - Assess the efficacy and safety of burosumab in infants and adolescents |
| - Assess the long-term efficacy and safety of burosumab in patients with XLH in terms of critical outcomes such as growth, bone shape, physical activity, tooth mineralization, hearing function, neurosurgical complications, and prevention of pseudofractures, enthesopathy, dental abscess and osteoarthritis |
| - Define the patients who will benefit most from burosumab treatment and, therefore, who should preferably initiate or switch to this treatment. For this, response predictors must be studieda |
| - Define the optimal dose and frequency of burosumab treatment for use once patients have reached disease stability |
| - Evaluate the cost-effectiveness of burosumab and conventional treatment in paediatric and adult patients with XLH |
| - Carry out bone quality studies with burosumab, evaluating the possible usefulness of bone turnover markersa |
In addition to the areas for research identified in the guidelines, this expert group considers that further research on the impact of this disease in terms of mortality should be undertaken. There are studies on the burden of disease that show an important impact in these patients in adulthood, especially in terms of persistence of pain, functional impairment and quality of life6,9,10,41, but little is known about the mortality associated with this condition. In a recent UK study of 122 possible cases of XLH, 9 (7.4%) had died with a median age of 64 years11.
Although, as the original guideline notes, more data are needed on long-term treatment with burosumab. Results from studies of at least 48 weeks of follow-up in both adults and children have recently been published. Results in adults show that burosumab continues to be well tolerated for up to 48 weeks and is able to sustainably correct phosphate levels, achieving fracture and pseudofracture consolidation and steadily improving some musculoskeletal disorders17. In children, results from clinical trials of 64 weeks duration show improvement in rickets severity, growth and biochemical abnormalities, with a favourable safety profile12,16; some case series in children with XLH treated with burosumab in clinical practice have shown results consistent with those described42.
We believe that another aspect not highlighted in the guidelines that should be further researched15 is the impact of burosumab on bone quality, evaluating the possible usefulness of bone turnover markers and techniques that non-invasively study other aspects of bone microarchitecture and strength, such as the Trabecular Bone Score (TBS) and, especially, high-resolution peripheral quantitative computed tomography (HR-pQCT)43. Recently a study in adult patients with XLH showed a significant increase in markers of bone resorption and bone formation after 48 weeks of treatment (77% increase in amino-terminal propeptide of procollagen type 1 [P1NP] and 36% increase in carboxy-terminal telopeptide of type I collagen [CTX])15.
Finally, we consider that more research is needed on vascular abnormalities in these patients, such as hypertension, ventricular hypertrophy and vascular calcification28,44, as well as disease-related residual osteoarticular involvement in adulthood6.
ConclusionsXLH is a rare genetic disease of phosphate metabolism that should be suspected in the following situations:
- 1
Clinical and/or radiological signs of rickets/osteomalacia associated with low age-adjusted serum phosphorus values with evidence of renal phosphate wasting (decreased TRP or TmP/GFR)
- 2
Angular deformity (malalignment) in the lower limbs, short stature and/or dental abnormalities associated with the previous point.
- 3
First-degree relatives with XLH
Other biochemical findings may include: increased alkaline phosphatase, increased FGF-23 (or inappropriately normal levels), slightly increased PTH levels or at the upper limit of normal, and low 1,25(OH)2 vitamin D levels (or inappropriately normal).
If available, testing for mutations in the PHEX gene and genetic counselling, in case of positive results, are advisable.
The conventional treatment in children (with phosphate supplements and active vitamin D adjusted according to age and clinical response) was until now essential to optimise growth and prevent as far as possible bone deformities and improve dental health. Until now, treatment in symptomatic adults (musculoskeletal pain, pseudofractures, dental problems) had to be done with oral phosphate supplements in divided doses and active vitamin D. The goal of treatment is to improve osteomalacia and related symptoms, as well as prevent dental complications (periodontitis, dental abscesses). Multidisciplinary follow-up is very important in these patients to ensure a comprehensive care for their different needs. The availability of new drug therapies targeting FGF-23, such as burosumab, represents a major breakthrough in the management of these patients. Trials and studies conducted with burosumab so far show clinical, laboratory and radiological improvement, with fewer side effects and better tolerability.
FundingThis project was funded by Kyowa Kirin Pharmaceuticals, S.L.U. Kyowa Kirin Pharmaceuticals has had no role in the evaluation of the guidelines, in the development of the recommendations, or in the preparation of this manuscript.
AuthorshipAll authors have contributed equally to the development of this project and the preparation of this manuscript; therefore, they are listed alphabetically.
Conflict of interestsDomingo González-Lamuño has received fees as a speaker from Kyowa Kirin Farmacéutica. Ana Lorente Rodriguez has received consultancy fees from Kyowa Kirin Farmacéutica. Silvia Marín-del Barrio has acted as a consultant, received speaker's fees and received training and scientific conference support from Kyowa Kirin Farmacéutica. Guillermo Martínez Diaz-Guerra has received fees for presentations and consultancy from Kyowa Kirin Farmacéutica. Pilar Peris has received speaker's fees from Amgen, Lilly, UCB and Kyowa Kirin Farmacéutica.
We wish to thank Fernando Rico-Villademoros (APICES, Madrid, Spain) for his editorial assistance regarding the preparation of this manuscript. This support has been funded by Kyowa Kirin Farmacéutica, S.L.U.
The following is Supplementary data to this article:
