





Paget’s disease of bone (PDB) is characterised by the alteration, in one or several bone locations, of the equilibrium between bone formation and bone resorption. This imbalance results in a disorganized, widened bone, in many cases with increased bone density, although more fragile. A genetic predisposition for PDB could explain between 5 and 40% of the cases. Different environmental factors should explain the rest of the cases. PDB was classically considered the second most common metabolic bone disease. However, in recent decades there has been a marked decrease in both incidence and clinical severity. These changes have led to believe that the influence of some environmental factor may have diminished or even disappeared. This decrease in incidence should not be an excuse for abandoning PDB research, but rather it should be the reason to remain searching to try to understand better its pathogenesis.
La enfermedad ósea de Paget (EOP) se caracteriza por la alteración, en una o varias localizaciones óseas, del equilibrio entre formación y resorción ósea. Este desequilibrio da lugar a un hueso ensanchado, desorganizado, en muchos casos con una densidad ósea aumentada, aunque más frágil. Existiría una predisposición genética para su desarrollo, que explicaría entre un 5 y un 40% de los casos, sobre la que actuarían distintos factores ambientales. La EOP fue considerada clásicamente la segunda enfermedad metabólica ósea más frecuente. Sin embargo, durante las últimas décadas presenta un marcado descenso tanto de la indecencia como de la gravedad clínica, lo que ha llevado a especular en la disminución o desaparición de la influencia de algún factor ambiental. Este descenso en la incidencia no debe servir como excusa para el abandono de su estudio sino ser la razón para tratar de entender mejor su patogenia.
Paget’s disease of bone (PDB) is the second most common disease of bone metabolism after osteoporosis. The disease is characterised by the appearance of bone foci in which there is an imbalance between bone formation and bone resorption, which accelerates bone replacement processes. This abnormality results in deformed areas of bone, with a disorganised pattern, increased vascularisation and increased fragility despite the apparent increase in bone mineral density.
The disease was first described by Sir James Paget in 1877 as osteitis deformans.1 Today, many patients with PDB are detected incidentally by imaging techniques that show bone lesions, or by the detection of an isolated elevation of alkaline phosphatase (ALP). Pain, bone deformity, osteoarthritis, fractures or deafness are the most common signs and symptoms.2
AetiopathogenesisCellular pathologyBone is constantly remodelling with a coupling between bone resorption and bone formation. Osteoclasts (derived from precursors of the macrophage lineage) are responsible for bone resorption. Following their action, osteoblasts (derived from precursors of the mesenchymal stem cell lineage) will form new bone. Some of these osteoblasts become included in the bone matrix, transforming into osteocytes which play a role in regulating the activity of osteoblasts and osteoclasts through the production of sclerostin and receptor activator for nuclear factor κ B ligand (RANKL).
In PDB we find osteoclasts increased in size and number and with more nuclei than normal osteoclasts, with a greater resistance to aeoptosis.3 They are more active and present nuclear inclusions4 whose function is unknown. A viral origin has been speculated, although it is currently postulated that they could be aggregates of undegraded proteins due to defects in autophagy.5 This activation pathway involves proteins that are also involved in autophagy processes, indicating that it may be an alteration of this pathway that leads to the alterations in PDB osteoclasts. The best known among these is p62, which is also associated with a gene related to susceptibility to developing PDB. This p62 protein plays a crucial role in recruiting ubiquitinated proteins to be eliminated in the autophagosome. Animal models of PDB show increased levels of p62 in hepatic osteoclasts.6 Optineurin (OPTN) or valosin-containing protein (VCP) also play a role in autophagy. Whether these defects in autophagy are the direct cause of increased osteoclast formation or simply a collateral phenomenon due to the involvement of proteins involved in both osteoclastogenesis and autophagy (p62, OPTN, VCP) is not yet known.
Genetic predispositionThe first description of PDB as a possible hereditary disease due to its high prevalence of familial cases was published in 1949.7 The presence of this familial aggregation is quantified as a 7–10 times higher risk of developing the disease in first-degree relatives of affected patients than in the general population.8This familial aggregation, together with the rarity of the disease in non-Caucasian populations9 and an increased number of PDB cases in certain rural areas (hotspots of high prevalence),10 indicates the existence of a predisposition to the development of the disease determined by genetic factors.
In recent decades, several mutations have been identified that belong to loci close to genes involved in the interaction between bone cells (Fig. 1). The identification of these genes has been made possible by genetic linkage analysis and genome-wide association studies (GWAS).
 is key to differentiation from osteoclast/macrophage lineage stem cell to osteoclast. Fusion of osteoclast precursors to form mature osteoclasts requires dendritic cell specific transmembrane protein (DC-STAMP). For osteoclast differentiation and activation, activation of the receptor activator for nuclear factor κ B ligand (RANK) is required. The union of the ligand of the receptor activator for nuclear factor kappa B (RANKL), released by the osteoblasts, with RANK, present in the cell membrane of the osteoclasts, gives rise to the activation of the nuclear factor kappa light chain enhancer in the B cells (NFκB). It remains in its inactive state in the cell cytosol forming a complex with the IBκα inhibitory protein. The binding of RANKL and RANK results in the activation of IκB kinase (IKK). This kinase phosphorylates the IκBα protein leading to ubiquitination and dissociation of the NFκB+IκBα complex. The activated NFκB enters the nucleus, binding to deoxyribonucleic acid (DNA) and leading to the activation of messenger ribonucleic acid (RNA) and protein formation resulting in a change in cellular function. RANK is also involved in the regulation of autophagy. Interaction with p62 is key to the activation of NFκB, facilitating translation of the RANK signal. Tumour necrosis factor 6 receptor (TRAF6) and CYLD (conserved cylindromatosis) are involved in activation. Osteoprotegerin (OPG) is a soluble protein with a high structural similarity to RANKL, also released by osteocytes, which competes with RANK in its binding to RANK and down-regulates NFκB activation by preventing RANKL-RANK binding. Valosin-containing protein (VCP) and optineurin (OPTN) are involved in the regulation of NFκ B signalling.")
Regulation of osteoclast physiology. Macrophage colony-stimulating factor (M-CSF) is key to differentiation from osteoclast/macrophage lineage stem cell to osteoclast. Fusion of osteoclast precursors to form mature osteoclasts requires dendritic cell specific transmembrane protein (DC-STAMP). For osteoclast differentiation and activation, activation of the receptor activator for nuclear factor κ B ligand (RANK) is required. The union of the ligand of the receptor activator for nuclear factor kappa B (RANKL), released by the osteoblasts, with RANK, present in the cell membrane of the osteoclasts, gives rise to the activation of the nuclear factor kappa light chain enhancer in the B cells (NFκB). It remains in its inactive state in the cell cytosol forming a complex with the IBκα inhibitory protein. The binding of RANKL and RANK results in the activation of IκB kinase (IKK). This kinase phosphorylates the IκBα protein leading to ubiquitination and dissociation of the NFκB+IκBα complex. The activated NFκB enters the nucleus, binding to deoxyribonucleic acid (DNA) and leading to the activation of messenger ribonucleic acid (RNA) and protein formation resulting in a change in cellular function. RANK is also involved in the regulation of autophagy. Interaction with p62 is key to the activation of NFκB, facilitating translation of the RANK signal. Tumour necrosis factor 6 receptor (TRAF6) and CYLD (conserved cylindromatosis) are involved in activation. Osteoprotegerin (OPG) is a soluble protein with a high structural similarity to RANKL, also released by osteocytes, which competes with RANK in its binding to RANK and down-regulates NFκB activation by preventing RANKL-RANK binding. Valosin-containing protein (VCP) and optineurin (OPTN) are involved in the regulation of NFκ B signalling.
The search for candidate genes to explain the aetiology of PDB by linkage analysis in families with inherited PDB was most successful at the turn of the century with the publication of a study demonstrating the presence of mutations on chromosome 5q35 in French-Canadians families11 which was replicated in UK families.12 This locus is related to the sequestosome-1 gene (SQSTM1) encoding the p62 protein. Subsequent studies have detected up to 30 other mutations related to the SQSTM1 gene. Although mutations in the SQSTM1 gene currently remain the main identified genetic cause of PDB, their spectrum is not universal and is detected in only 25–40% of familial cases and in less than 10% of patients without them.13 These mutations in the MQTS1 gene are considered to be germline stem cell mutations, i.e., they affect all cells in the body that are descendants of the original mutated cell. In addition to this germline genetic inheritance, somatic mutations (mutations found only in cells of the affected tissues) in the MQTS1 gene have also been described in cases of sporadic PDB.14,15 This may partly explain the focal nature of PDB.
In addition to PDB, linkage analysis in other inherited diseases related to the PDB spectrum have demonstrated mutations related to bone metabolism. These include: 1) familial expansile osteolysis (OMIM 174810): TNFRSF11a gene associated with alterations in RANK signaling,16 2) juvenile Paget's disease (OMIM 239000): TNFRSF11b gene encoding osteoprotegerin17 and 3) PDB, frontotemporal dementia and inclusion body myopathy complex (OMIM 167320): associated with the VCP gene.18 The linkage analysis in PDB did not show association in the case of the TNFRSF11a genes19 (although they would show association in GWAS as will be seen later, and in studies using single nucleotide polymorphisms20) or in VCP,21 but they did show association in the case of TNFRSF11b.22
GWAS have identified 7 additional loci that would explain 13% of the risk in patients with PDB and a family history who do not have mutations in the SQSTM1 gene.23,24 The genes related to the physiology of the osteoclast stand out: (a) the CSF1 gene, encoding macrophage colony stimulating factor, (b) TNFRSF11A, encoding RANK, (c) TM7SF4, encoding dendritic cell-specific transmembrane protein, (d) OPTN encoding optineurin or (e) RIN3, encoding a guanine exchange factor expressed by osteoclasts (Fig. 1). The genes involved in the other identified loci are less clear. The presence of any of these 7 additional loci increases the risk of developing the disease by 1.4 to 1.7. Finally, the possible association of PDB with other mediators of osteoclast activity such as proinflammatory cytokines25 has also been studied.
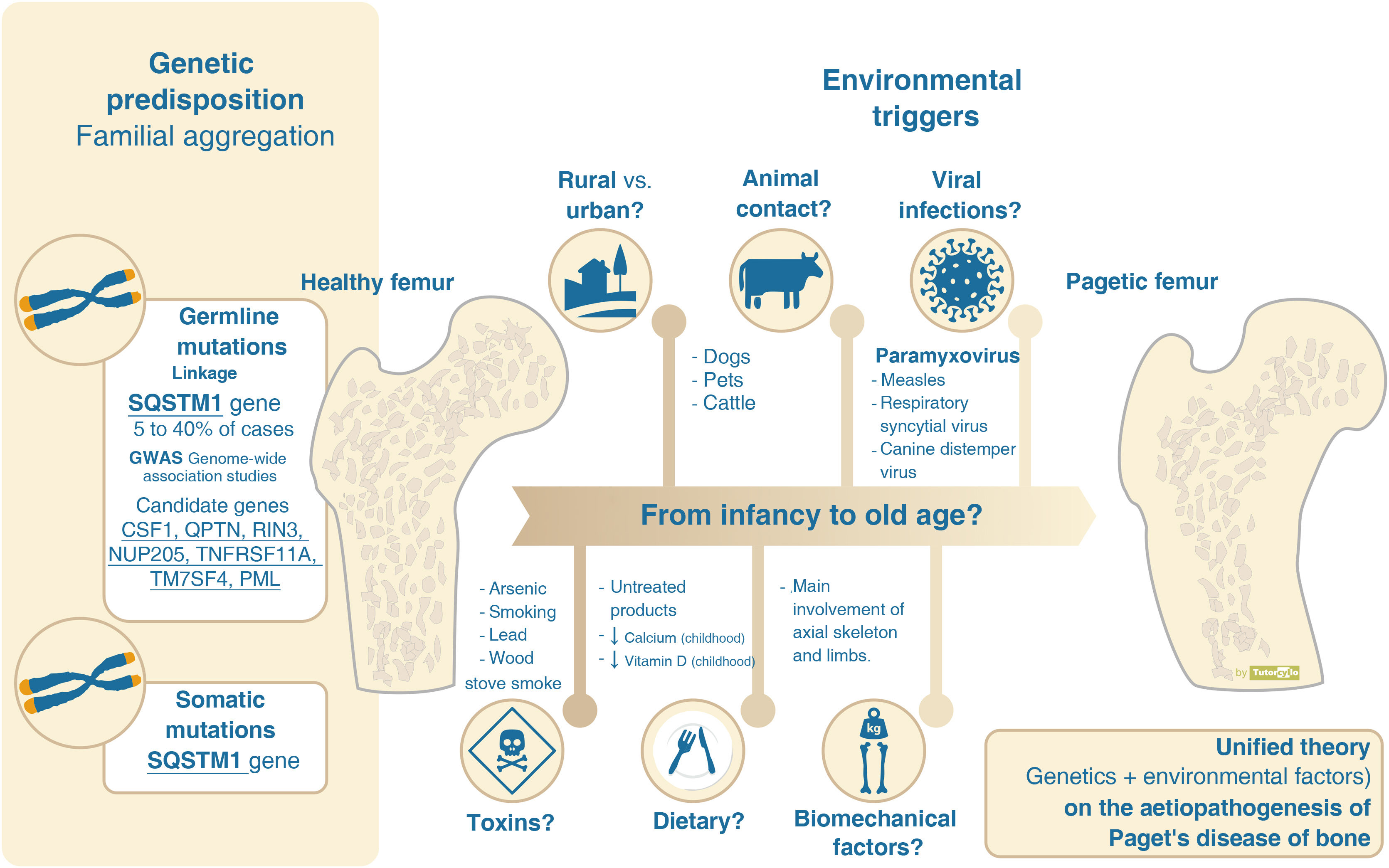
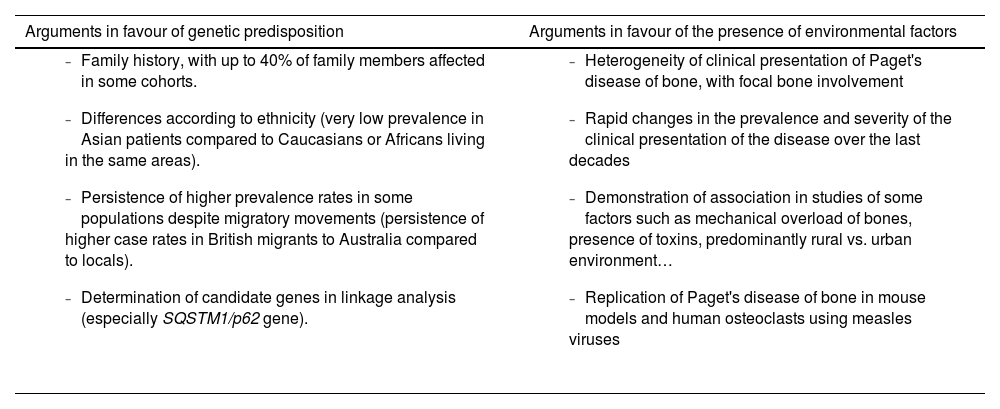
However, the hereditary component is not sufficient to explain the disease and there is evidence supporting the need to consider the interaction with environmental factors (Table 1). The decline in incidence and severity described in recent decades10 would favour the environmental theory. This decline has even been demonstrated in SQSTM1 gene mutation carriers. These patients now have a more attenuated phenotype than their progenitors and have a later age of diagnosis than their progenitors.26,27 The reason for this delay in genetic penetrance is unknown. Decreased exposure to environmental factors is suggested as the most likely mechanism to explain these phenomena (Fig. 2).
Aetiology of Paget’s disease of bone. Predisposition versus influence of environmental factors.
| Arguments in favour of genetic predisposition | Arguments in favour of the presence of environmental factors |
|---|---|
|
|

Aetiopathogenesis of Paget’s disease of bone, unified theory. Several genes have been implicated in the aetiopathogenesis of Paget's disease of bone. However, this genetic component cannot explain more than 40% of familial forms and less than 10% of sporadic forms. The presence of yet unconfirmed environmental factors must be necessary for the development of the disease.
Several possible environmental factors may be involved in the pathogenesis of PDB, although there is no clear evidence for any of them.
One of the most documented environmental factors is the involvement of viral infections. The presence of inclusions in osteoclasts reminiscent of paramyxovirus-related particles28 has led to the hypothesis of viral aetiology. Evidence for the origin of these inclusions is conflicting, with the initial demonstration of viral particles in osteoclasts29 or in circulating blood cells30 not being replicated in subsequent studies.31 There is no agreement on their role. Nor has the analysis of antibody response to different viruses (paramyxoviruses: measles, respiratory syncytial virus, canine distemper virus, mumps, rubella or varicella zoster) been associated with PDB or its severity.32 The elicitation of PDB-like changes in laboratory or mouse models by infusion of measles virus or its nucleocapsid supports the theory of viral origin.33
Other factors with which PDB has been linked are: 1) contact with animals such as dogs,34 not confirmed by other researchers,35 2) residence in rural areas,36,37 3) dietary factors such as low milk intake38 or vitamin D deficiency39 in childhood, 4) the presence of toxins such as arsenic,40 smoking41 or the use of wood stoves in childhood,42 or 5) biomechanical factors, as the bones affected are mainly those related to loading, as in the case of the snooker player and the pagetic involvement of the hand.43
In any case, the theory that seems most feasible in order to understand the complexity of the aetiopathogenesis of PDB is that there may be a genetic susceptibility associated with the participation of different, as yet unconfirmed, environmental factors that lead to the development of the disease over the course of years. This theory is known as the unified theory. These environmental factors, in addition to acting on patients with a genetic predisposition, could also lead to local epigenetic changes in other patients without such a predisposition.44 These epigenetic changes could account for the heterogeneous presentation of the disease in each patient, although this has not yet been demonstrated. Finally, the role of microRNAs, small non-coding RNA strands that regulate gene expression, including genes related to bone metabolism and osteoclast formation, also seems to be a future line of research.45
The ZiPP study,46 where healthy subjects who are relatives of PDB patients and carry mutations in the SQSTM1 gene, undergo several years of follow-up, will provide further insight into the role of the presence of genetic predisposition associated with mutations in SQSTM1 in the development of the disease. In this study, healthy subjects were randomised to receive a dose of zoledronate or placebo at the start of follow-up.
EpidemiologyAs PDB is a bone disease, and therefore leaves a lasting trace over time, it is interesting to refer to archaeological studies to discuss the epidemiology of the disease. The high prevalence of the disease in the United Kingdom and in countries with a high proportion of Anglo-Saxon-origin population, such as the former colonies of Australia or New Zealand, has led several authors to propose the British Isles as the place of origin of the disease. This seems to be confirmed by a significant increase in the number of skeletons with PDB traits from the UK compared to other European countries.47 This theory is refuted in a Spanish study which reviews the described palaeopathological cases of the disease, with the oldest described in French, Italian or Spanish sites, so the authors postulate that the disease did not originate in Britain but was probably introduced to the islands during the Roman occupation.48 Furthermore, these authors describe cases found in pre-Columbian America sites, which makes the true origin of the disease more uncertain.
The epidemiology of PDB has been characterised by a significant heterogeneity between different areas of the world, with a clear increase in prevalence in areas of Anglo-Saxon origin and under-representation in Asia or Africa. In addition to the significant difference between countries, there is also great disparity within countries, with the presence of high prevalence PDB areas in regions where the number of cases is much higher than in neighbouring regions. The best known high-prevalence area is Lancashire,49 in the UK. High-prevalence pockets have also been described in Italy and Spain, such as Vitigudino in Salamanca,50 or Sierra de la Cabrera in Madrid.51 The Spanish and Italian high-prevalence areas are characterised by being rural, sparsely populated and somewhat isolated areas. When attempts have been made to explain the origin of these clusters, the presence of environmental factors, including those related to contact with animals, has been considered. In the case of Italy, there is a high proportion of familial forms, indicating a genetic origin.
However, if anything makes the epidemiology of PDB interesting, it is not its heterogeneity, but the significant changes it has undergone in recent decades,10 with an extreme decline in prevalence, even more significant in high-prevalence areas.52 This decline has been accompanied by a delay in the presentation of the disease, which is being diagnosed later and later, and a decrease in the severity of the presentation, with less extensive skeletal involvement and lower laboratory values. Although attempts have been made to explain part of these changes by differences in genetic background due to significant immigration from areas of low PDB prevalence in recent decades in countries such as the United Kingdom (from India or Pakistan) or Spain (from South American countries), the fact is that this immigration has not significantly affected the rural areas such as Vitigudino, which have been the areas where the decline in the prevalence of PDB has really been most noticeable. Reduced exposure to some yet-to-be-determined environmental agent, such as changes related to veterinary livestock control, may better explain the drastic changes in the prevalence of PDB.
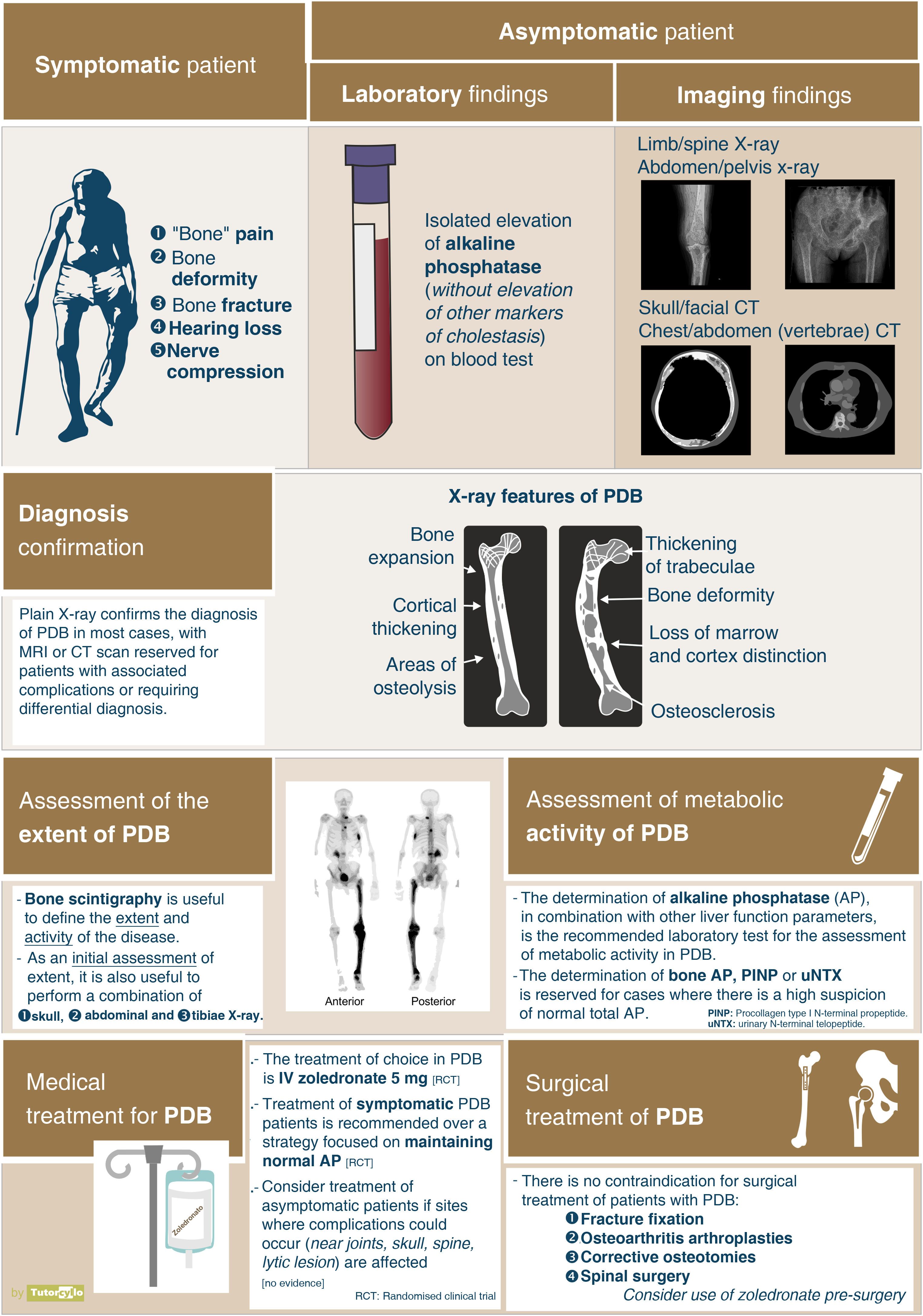
Clinical manifestationsA systematic review published in 20142 assessed the clinical presentation of the disease. The main symptom of patients with PDB was disease-related pain, although the prevalence of this symptom varied very significantly between the different series included in the review (from 18% to 92% of cases), making it difficult to estimate a true prevalence of symptoms. These differences are also confirmed in our setting. The general feeling is that the vast majority of patients are asymptomatic, and their diagnosis is made incidentally when other diagnostic tests (imaging tests or AP determination) are performed. In a recently published British series, up to 74% of patients with PDB diagnosed in primary care were not referred to specialist consultations to assess treatment.53
In addition to the pain caused by the local disease activity itself, the deformity caused by the disease leads to osteoarthritis of the joints, which also generates local pain. Differentiating between pain caused by the activity of the disease and pain secondary to osteoarthritis resulting from the deformity to which it has given rise is often complex and will lead to the establishment of antiresorptive treatments that will not improve the patient's clinical condition.
Bone deformity also leads to neurological disorders, due to compression of the nerves or stenosis of the spinal canal, and deafness, due to alteration of the bony structures of the inner ear.
Fractures of affected bones are also increased as these bones are poorly structured and are less able to withstand biomechanical loads.
Classic manifestations such as heart failure associated with increased vascularisation of the bone, hypercalcaemia or neoplastic transformation of the lesions into osteosarcomas or giant cell tumours54 are now uncommon or even non-existent.
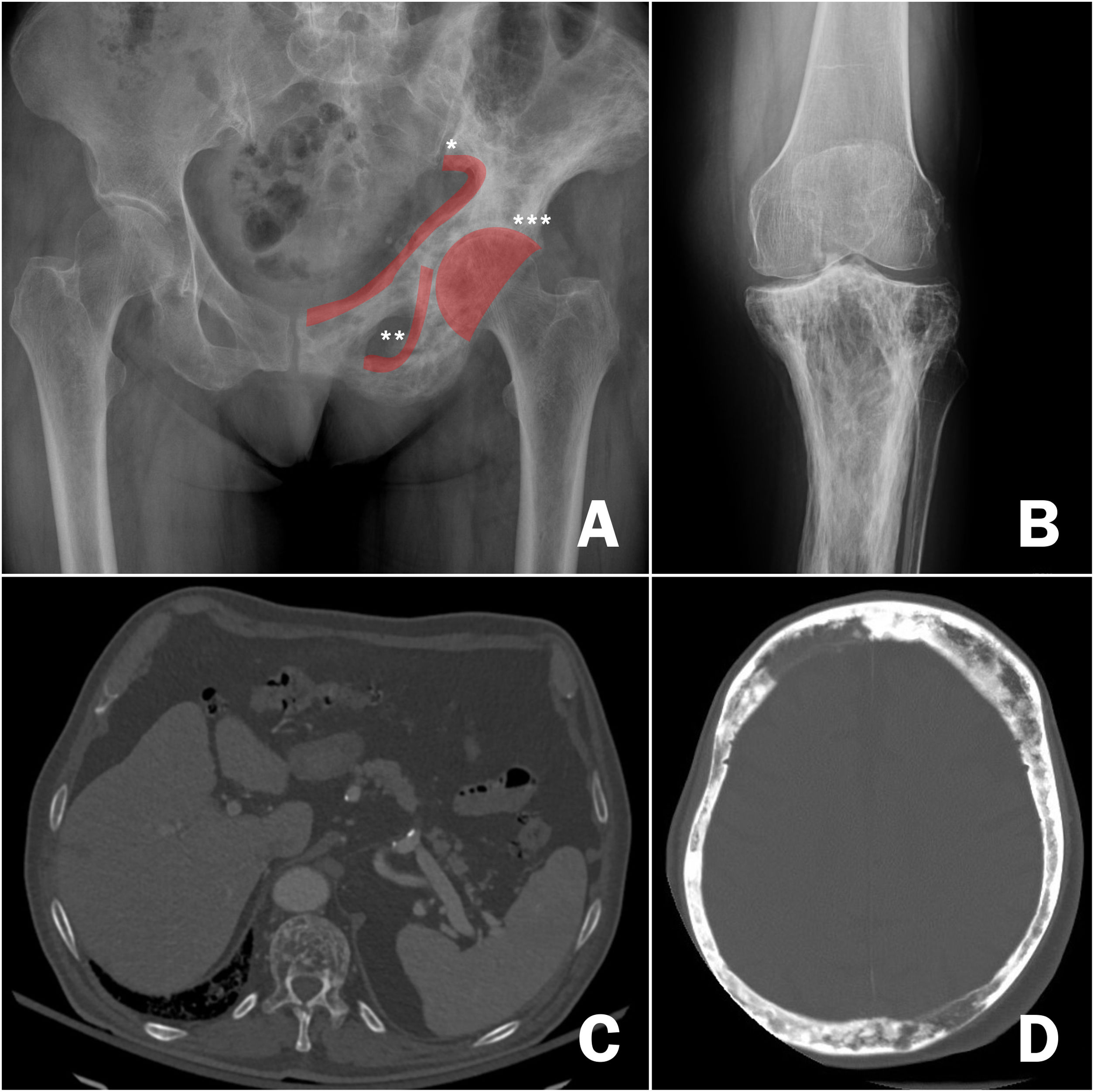
DiagnosisOnce the suspicion of PDB has been established, it is necessary to confirm it by performing imaging tests to demonstrate the changes inherent to the disease (Fig. 3). In the first phase of bone involvement, a "lytic front" or "Blade of Grass" is described, with cortical involvement. Subsequently, the pagetic bone will be characterised by the presence of both a thickening of the cortex and an altered and coarse trabeculation that eventually leads to enlargement and deformity of the bone (Fig. 3B, comparison between the cortex and trabeculae of the healthy femur and the tibia with PDB). The association of osteoblastic and osteolytic areas in the disease is characteristic. In cases of spinal involvement, PDB lesions manifest with increased opacity, as do osteoblastic metastases of some tumours. However, the deformity of the vertebra and the increased cortical thickness often make differentiation between the two easy. In other locations such as the pelvis, it will also be relatively easy to differentiate osteoblastic involvement of PDB from metastatic tumours, since the bone is enlarged in PDB, the iliopectineal and ilioischial lines are altered or the acetabulum is deformed (Fig. 3A). In cases of diagnostic doubt or suspected complications, it is advisable to request additional diagnostic imaging studies such as computed tomography or magnetic resonance imaging. Only in cases where, despite these imaging tests, it is not possible to establish the diagnosis of the disease, a bone biopsy should be requested. The need to carry out a pathological examination for the confirmation of PDB is very rare in routine clinical practice.

Radiological images of Paget's disease of bone. A) Involvement of the left hemipelvis, with alternating osteolytic and osteoblastic images and thickening of the cortices, *iliopectineal line, ** ilioischial line, *** acetabulum. B) Involvement of the upper tibia, with significant cortical thickening and alteration of bone trabeculae in the upper regions. C) Involvement of the thoracic vertebrae on pulmonary computed axial tomography, highlighting the alteration of the structure of the vertebral body. D) Cranial involvement in cranial computed axial tomography requested for stroke. Significant cortical thickening.
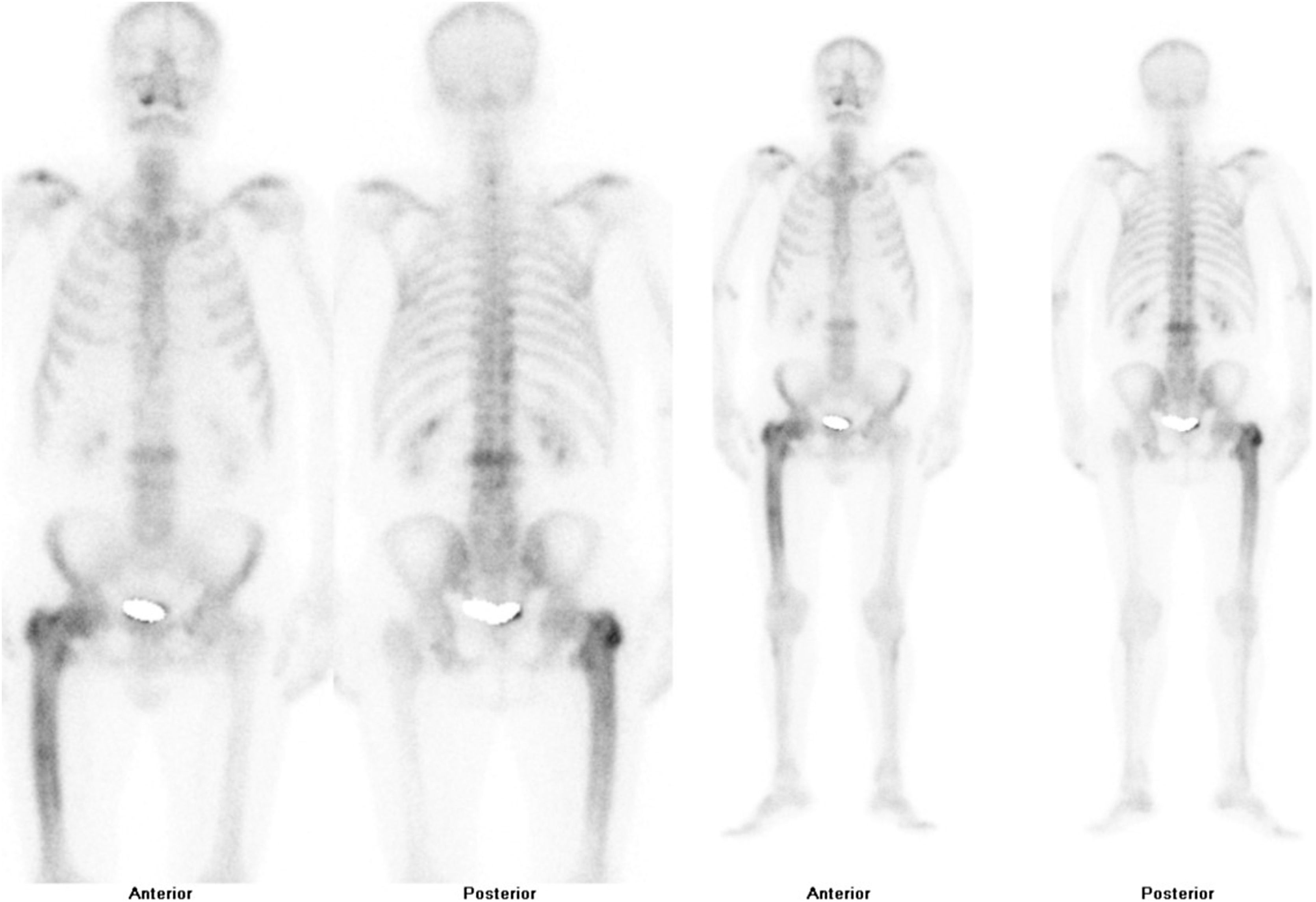
After confirmation of the disease on plain X-ray or computed axial tomography, the study of the extent of the disease is completed by assessing the possibility of involvement in other areas of the skeleton. Technetium-99 bone scintigraphy (Fig. 4) is the most commonly used test. An alternative strategy would be to perform a radiological bone series including the skull and face, abdomen and tibiae. This approach demonstrated a sensitivity of 93% in a Spanish PDB cohort.55

Regarding the assessment of the biochemical activity of the disease, the determination of AP is the most standardised test, with the detection of its isolated elevation within the determinations of hepatic cholestasis. Only in the case of high suspicion with normal AP it might be useful to request additional bone turnover markers such as the bone fraction of AP or the procollagen type 1 N-terminal propeptide or the N-terminal telopeptide of collagen type 1.
TreatmentThe main indication for treatment is the control of local pain in relation to an affected area in cases where it is related to the disease activity itself. The use of bisphosphonates is effective in reducing pain in these patients, with a required number of patients to treat of 5, with a confidence interval between 1 and 35.56 The bisphosphonate of choice is intravenous zoledronate, which has been shown to be superior to other bisphosphonates such as risedronate57 or pamidronate. Other treatments such as calcitonin may help with pain control, but its use is currently limited to patients who are contraindicated for bisphosphonate administration. There are published cases of PDB response with the use of denosumab, another antiresorptive drug, but it is not currently approved for use in this indication.
Intravenous zoledronate treatment can cause flu-like symptoms in the first 24−48h, with myalgia, headache or nausea in almost one in 4 patients during the first infusion, less common in subsequent infusions. This condition is self-limiting. Some authors suggest the use of prophylactic paracetamol or dexamethasone58 on the day of treatment and thereafter. It will also be necessary to know the patient's vitamin D levels before administration since there is a risk of hypocalcaemia after administration of the drug in patients with hypovitaminosis.
Although the indication for treatment in patients with PDB and pain in the affected site is established, the difficulty that we will encounter in many patients will be to find out the origin of the pain. Identifying whether it is caused by the activity of the PDB itself, or other causes related to it, such as osteoarthritis determined by bone deformity, nerve deformity-related compressions or pseudofractures, will not always be easy. In these cases, the patient’s medical record and a clinical examination will provide us with the most appropriate information to decide the origin of the pain. The presence of elevated bone formation markers, as in the case of AP, can help in decision-making, since disease activity is usually associated with elevated AP levels, but should not direct the decision. In cases where the origin of the pain is unclear, a therapeutic test with intravenous zoledronate administration may be chosen. When the pain improves, it may be attributed to disease activity; when there is no pain relief, further evaluation of the patient should be considered to identify the cause of the pain.
In general, the treatment of PDB is considered in patients in whom bone involvement has already been established, with bone alteration and deformity, with the consequent sequelae for the patient. There are currently no medical treatments that allow for the reversal of bone changes, so the usefulness of treatment in preventing progression to osteoarthritis, reversing bone deformity, improving hearing loss in the case of skull involvement or alleviating nerve compression related to bone deformity is unclear. There is also no evidence regarding the influence of the treatments on the patient's quality of life. In all these cases, if there is no pain, there is no clear indication for treatment. It is under debate whether the presence of asymptomatic but biochemically active PDB with lesions in bone sites where there is a risk that the disease could lead to complications (near the joints, in the skull, in the spine, with the presence of large lytic foci) could be a reason for treatment despite the lack of evidence on the usefulness of treatment. The PRISM-EZ clinical trial59 compared 2 therapeutic strategies in PDB: a) guiding treatment with the aim of controlling the patient's pain or b) guiding treatment with the aim of controlling elevated bone formation (AP) markers. The strategy aimed at controlling elevated AP did not benefit patients, as it tended to increase the number of fractures and trauma surgery without improving either pain control or quality of life. The goal of treatment of patients with PDB should be symptom control, without being obsessed with achieving a suppression of biochemical bone turnover markers. There is therefore no set administration interval for bisphosphonates, as for many patients a single dose will be sufficient, while others will need to repeat the dose months or years after the initial dose.
In addition to pharmacological treatments, orthopaedic and traumatological surgery may be necessary in patients with PDB due to the presence of fractures, bone deformities, osteoarthritis or nerve compression. There is no contraindication in these patients for trauma surgery such as osteotomies, fracture fixation surgery, arthroplasty or spinal surgery for nerve decompression. Bone deformity and increased vascularisation of the affected bone will pose an additional challenge for traumatologists due to the technical difficulties that may arise. The results of different series point to a good result in arthroplasty and a good consolidation of the pagetic bone with the exception of the proximal femur. The use of intravenous zoledronate prior to surgery could reduce the risk of bleeding during surgery, although there is no evidence for this indication. In the case of the pre-surgical use of bisphosphonate, the greatest concern is the possible repercussion that the action of the drug could have on the consolidation of fractures. There is also no contraindication for the use of cochlear implants or hearing aids in patients with hypoacusis.
The clinical guidelines of the European and American societies (European Calcified Tissues Society and American Society of Bone and Mineral Research) establish a series of recommendations for the treatment of PDB.60Fig. 5 summarises some of the main recommendations.
Prognosis
The prognosis of PDB is variable. Many patients remain asymptomatic. In most patients with pain, a single lifetime administration of zoledronate results in pain improvement and control of AP levels. The results of the ZiPP study46 will show in the coming years whether the administration of zoledronate in healthy individuals with SQSTM1 mutations can alter the prognosis of the disease by preventing or delaying its onset.
FundingNo funding has been received for this article.
Conflict of interestNo conflict of interest is declared.
I wish to thank Dr. del Pino Montes, a recently retired internist and rheumatologist, for all his dedication to PDB. Dr. del Pino is a pioneering figure in the study of this disease in Spain. His contribution was key to the knowledge and characterisation of the high-prevalence area of Vitigudino, in the province of Salamanca, one of the best-known high prevalence foci of the disease in the world. Without his passion for this disease, this review would not have been possible.
