



La enfermedad por depósito monoclonal de inmunoglobulina (MIDD) es un tipo raro de paraproteinemia que se caracteriza por la presencia de depósitos monoclonales de inmunoglobulinas en la membrana basal glomerular, manifestándose más frecuentemente en la quinta y sexta década de vida. La MIDD se subclasifica en la enfermedad por depósitos de cadenas ligeras (LCDD), enfermedad por depósitos de cadenas pesadas (HCDD) y enfermedad por depósitos de cadenas ligeras y pesadas (LHCDD), siendo la LCDD la forma más frecuente de presentación en donde se identifican cadenas ligeras kappa hasta en el 80%, asociándose hasta el 50% de pacientes con mieloma múltiple. El hallazgo de esclerosis nodular se puede asociar a LCDD en la mayoría de pacientes.
Monoclonal immunoglobulin deposit disease (MIDD) is a rare type of paraproteinaemia that is characterized by the presence of monoclonal deposits of immunoglobulins in the basal glomerular membrane, manifesting more frequently in the fifth and sixth decade of life. MIDD is subclassified as light chain deposit disease (LCDD), heavy chain deposit disease (HCDD) and light and heavy chain deposit disease (LHCDD), with LCDD being the most frequent form of presentation where kappa light chains are identified in up to 80%, associated with multiple myeloma in up to 50% of. The finding of nodular sclerosis can be associated with LCDD in most patients.
La enfermedad por depósitos de cadenas ligeras es una entidad poco frecuente caracterizada por los depósitos monoclonales de cadenas ligeras no amiloidóticas a nivel renal. Reportamos el caso de una paciente con mieloma múltiple que presentó una pérdida acelerada de la función renal, hipocomplementemia y proteinuria no nefrótica como manifestación clínica de su enfermedad. En este caso los depósitos monoclonales de cadenas ligeras kappa se expresaron como lesiones tipo esclerosis nodular a nivel glomerular.
Exposición del casoPaciente mujer de 69 años hospitalizada por astenia, disnea progresiva, edema en miembros inferiores y orina espumosa de 6 meses de evolución; antecedente de hipertensión de 2 años. Diagnosticada de insuficiencia cardiaca 3 meses antes de su ingreso por congestión pulmonar. Ecocardiografía FEVI disminuida (FEVI: 37%), discinesia septal, inferior y anterior y disfunción diastólica restrictiva y efusión pericárdica moderada. En la tomografía pulmonar se evidenció hepatomegalia y poliserositis (derrame pleural bilateral, líquido ascítico y derrame pericárdico).
Al ingreso cursó con deterioro de la función renal. Al examen físico había edema generalizado, palidez, PA 110/50mmHg. En el laboratorio, creatinina al ingreso de 0,9mg/dl y se incrementó hasta 6,9mg/dl durante su hospitalización, Hb 9,7g/dl, albúmina 3,2g/dl, globulina 2,1g/dl, calcio 9,4mg/dl. Proteinuria de 24h en 2,1g, sedimento urinario: hematíes 0-2×C, serología para hepatitis B y C, sífilis y VIH, negativa; marcadores inmunológicos anticuerpos anticitoplasma de neutrófilos (ANCA) negativo, anticuerpos antinucleares (ANA) con prueba de inmunofluorescencia indirecta (IFA) 1/100 patrón nucleolar, anticuerpo contra antígenos nucleares extraíbles —del núcleo de células en proliferación (ENA-PCNA) (++)—, complemento C3 disminuido (58,1mg/dl) y C4 normal. La electroforesis en suero no presentó pico monoclonal, y en orina tuvo elevación de la fracción gamma e inmunofijación con proteína de Bence Jones con componente monoclonal en cadenas ligeras kappa. Determinación de cadenas ligeras kappa 1.040mg/l, lambda 35mg/l y cociente kappa/lambda 29,3. Se realizó aspirado de médula ósea con resultado de 80% de células plasmáticas infiltrando la muestra recibida. La ecografía renal refirió riñones de tamaño conservado. No se objetivaron lesiones osteolíticas por radiología.
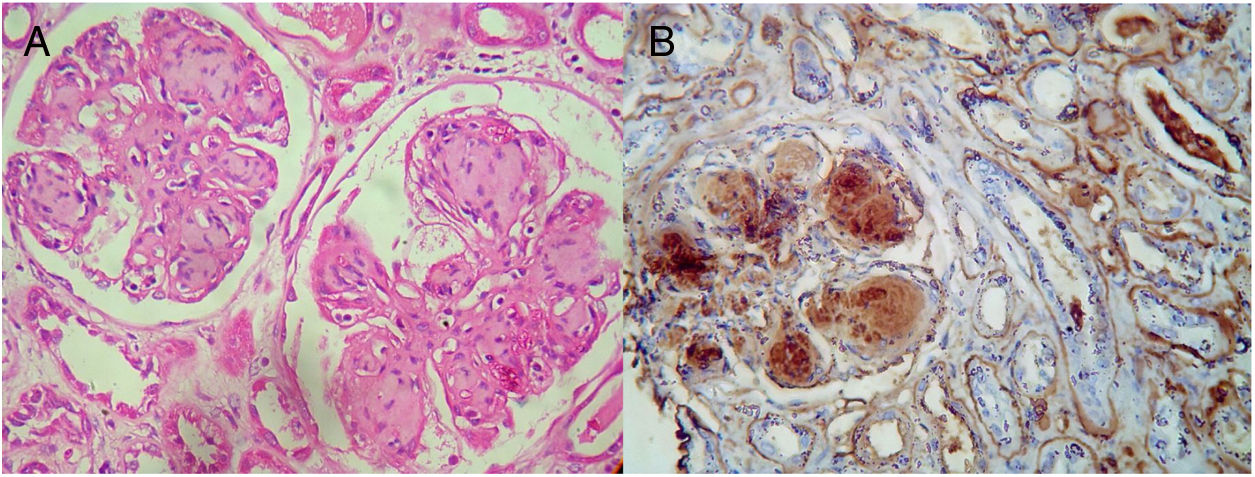
Se realizó la biopsia renal (fig. 1) constituida por 75 glomérulos, 5 globalmente esclerosados, los glomérulos viables con expansión mesangial formando nódulos hialinos acelulares. Los nódulos fueron PAS positivo, rojo de Congo negativo, intersticio con infiltración linfomononuclear leve. Inmunohistoquímica positiva para kappa. No hubo presencia de cilindros tubulares, siendo el compromiso glomerular.
 Expansión mesangial con nódulos hialinos acelulares (esclerosis nodular). B) Inmunohistoquímica kappa positivo 3+, lambda+/− y CD34 positivo.")
El diagnóstico fue de glomeruloesclerosis nodular por depósito de cadenas ligeras kappa asociado a un mieloma múltiple.
DiscusiónLa enfermedad por depósitos de cadenas ligeras se presenta más frecuentemente entre los 55 y 60 años de edad y se asocia con mayor frecuencia al mieloma múltiple, aunque puede asociarse a otras enfermedades linfoplasmáticas con una incidencia de aproximadamente el 5%1,2. Clínicamente, la proteinuria en rango nefrótico es común y el síndrome nefrótico se presenta en el 25% de pacientes. Los pacientes presentan usualmente proteinuria, hematuria microscópica, hipertensión y grados variables de insuficiencia renal3-5. Debido a las variadas presentaciones clínicas y múltiples alteraciones morfológicas, es posible que ambas entidades sean poco reconocidas y diagnosticadas. En este sentido, el reconocimiento de la enfermedad del presente caso atravesó por diversos diagnósticos diferenciales.
Las manifestaciones clínicas al ingreso del presente caso se caracterizaron por compromiso glomerular y cardiaco. Cuando la paciente fue diagnosticada de insuficiencia cardiaca no presentó insuficiencia renal, excluyendo entonces la posibilidad de un síndrome cardiorrenal, por lo que la oliguria y sobrecarga de volumen no constituyen factores causales. El compromiso cardiaco, renal, serosas e hipocomplementemia, en el contexto de una enfermedad glomerular, sugieren descartar enfermedades sistémicas como lupus eritematoso sistémico, específicamente una glomerulonefritis proliferativa difusa tipo IV; sin embargo, una serología para lupus por ELISA negativa, una titulación por IFA en 1/100 y la ausencia de hematuria alejan esta posibilidad; más aún cuando el compromiso de la función renal fue progresivo sin alteraciones analíticas compatibles para lupus. En este caso la prueba que intervino como cribado para la discrasia de células plasmáticas fue el proteinograma electroforético en orina, en donde hubo un pico en la fracción gamma y que fue debidamente detectado en la inmunofijación urinaria. El incremento marcado de las cadenas ligeras kappa en suero sugirió el descarte de mieloma múltiple, pese a que en este caso hubo ausencia de manifestaciones clínicas como dolor óseo, disproteinemia, hipercalcemia y pico monoclonal sérico. El diagnóstico de mieloma se confirmó por la presencia de plasmocitos clonales en la médula ósea asociados al compromiso renal. El compromiso renal por depósito monoclonal de cadenas ligeras por la paraproteinemia se evidenció en la biopsia renal y, en este caso, se presentó como esclerosis nodular. El compromiso a nivel de las serosas probablemente estuvo asociado también al depósito de las cadenas ligeras a nivel pleural y pericárdico; esto último habría condicionado el factor restrictivo de la insuficiencia cardiaca. La infiltración pleural y pericárdica por la paraproteinemia ha sido reportada en la literatura6,7.
La esclerosis nodular como hallazgo en la histopatología renal tradicionalmente presenta un diagnóstico diferencial con nefropatía diabética o amiloidosis, sin embargo, también se describe asociada a enfermedad por depósitos de cadenas ligeras en el 60% de los pacientes, compuesta frecuentemente por cadenas tipo kappa, los cuales pueden parecerse a los depósitos vistos en la nefropatía diabética nodular o en la amiloidosis de cadenas ligeras; sin embargo, este tipo de depósitos son rojo Congo negativo, no amiloidóticos y no presentan estructuras de tipo fibrilar en su ultraestructura8. En cuanto a la hipocomplementemia, se ha descrito que las proteínas monoclonales promoverían una inhibición de las proteínas reguladoras del complemento, favoreciendo la activación de la vía alterna del complemento; no obstante, su fisiopatología requiere aún de más evidencia9.
La supervivencia de la enfermedad por depósitos de cadenas ligeras a 5 años es del 70% aproximadamente, la cual se reduce notoriamente si se asocia a mieloma múltiple10. Los pacientes que presentan al momento del diagnóstico valores de creatinina sérica de más de 4mg/dl tienen peor pronóstico y alto riesgo de progresión a enfermedad renal crónica terminal2, por lo que es críticamente importante iniciar rápidamente el esquema de tratamiento para reducir las concentraciones de cadenas ligeras libres en pacientes con mieloma múltiple, puesto que dicha reducción se asocia con un aumento de la supervivencia. El régimen de tratamiento con bortezomib es favorable por su seguridad de uso en pacientes con insuficiencia renal, incluso en hemodiálisis8.
ConclusiónLa enfermedad por depósitos de cadenas ligeras fue descrita por primera vez hace más de 3 décadas. Presentamos un caso de glomerulopatía mediada por paraproteinemia en un paciente con diagnóstico de mieloma múltiple. Debido a las variadas presentaciones clínicas y los múltiples diagnósticos diferenciales y morfológicos, es posible que ambas entidades sean poco reconocidas y diagnosticadas. Conocer las diferentes formas de presentación puede ayudar en el diagnóstico precoz y tratamiento de la enfermedad.
FinanciaciónNinguna.
Conflicto de interesesNinguno.
