





Conferencias clínico-patológicas: la importancia del diagnóstico multidisciplinar
Más datosUn paciente de 71 años sin historia de reacciones adversas medicamentosas ni consumo de tóxicos. Natural de Huancavelica, procedente de Lima (Perú), que se encontraba en Barcelona visitando a unos familiares residentes en la ciudad. Trabajó como albañil y en una fábrica de cemento.
Como antecedentes patológicos relevantes el paciente reportaba informes médicos de su país de origen en los que se describía un episodio de polineuropatía (PNP) y mononeuritis catalogada de secundaria a vasculitis sistémica p-ANCA en relación a un episodio en 2010 de disminución de fuerza, hipostesias y dolor en los miembros inferiores. Consta la realización de un electromiograma que evidenció PNP sensitivo-motora crónica axonal de grado leve-moderado, con respuesta al tratamiento inmunodepresor, en ese momento con prednisona oral. También constaba de antecedente de fibrosis pulmonar no catalogada, de la que no se disponía de estudio de imagen.
Consultó en julio de 2017 en nuestro centro por un episodio brusco de hipostesias bilaterales ascendentes hasta la rodilla, acompañada de pérdida de fuerza progresiva en ambas piernas con imposibilidad para la deambulación. Exclusivamente se asoció una diarrea líquida autolimitada a las 24 h.
En la exploración se mostraba normotenso y afebril, eupneico en reposo con saturaciones del 94% en el aire ambiente. En la auscultación pulmonar se objetivaron crepitantes finos bibasales. En el examen neurológico no se objetivo afectación de los pares craneales ni asimetría o dismetrías. No había atrofia muscular, con fuerza y sensibilidad conservadas en las extremidades superiores a excepción de una leve hipoestesia bilateral en los dedos, en las extremidades inferiores presentaba disminución de fuerza distal en la extremidad inferior izquierda (3+/5) y ligera en la pierna derecha (4/5). Los reflejos osteotendinosos tricipital, bicipital y estiloradial eran normales, sin embargo, presentaba reflejos aquíleos ausentes. Además asociaba una hipoestesia bilateral por debajo de la rodilla con anestesia y disestesia en ambos pies. El reflejo cutáneo-plantar era indiferente de forma bilateral.
Se orientó el caso clínico como PNP sensitivo motora simétrica a estudio, y se realizaron diferentes exploraciones complementarias para la obtención de un diagnóstico diferencial.
El paciente presentaba unos valores séricos de glucosa, calcio, una función renal y un perfil hepático en rango de la normalidad. Analíticamente se descartó déficit de vitamina B12 y ácido fólico. Se objetivó un proteinograma con desplazamiento electroforético normal y una dosificación de inmunoglobulinas en rango. En relación con un posible desencadenante infeccioso, se realizaron serologías de virus hepatotropos B y C, virus de la inmunodeficiencia humana (VIH) y Borrelia con resultado negativo. En el estudio también se obtuvo un patrón de autoinmunidad en el que destacaba; ANA positivos a título 1/160 con patrón moteado y p-ANCA positivo a título de 1/120 acompañado de MPO de 5,8. Además de factor reumatoide positivo a título 478 y antipéptido cíclico citrulinado negativo.
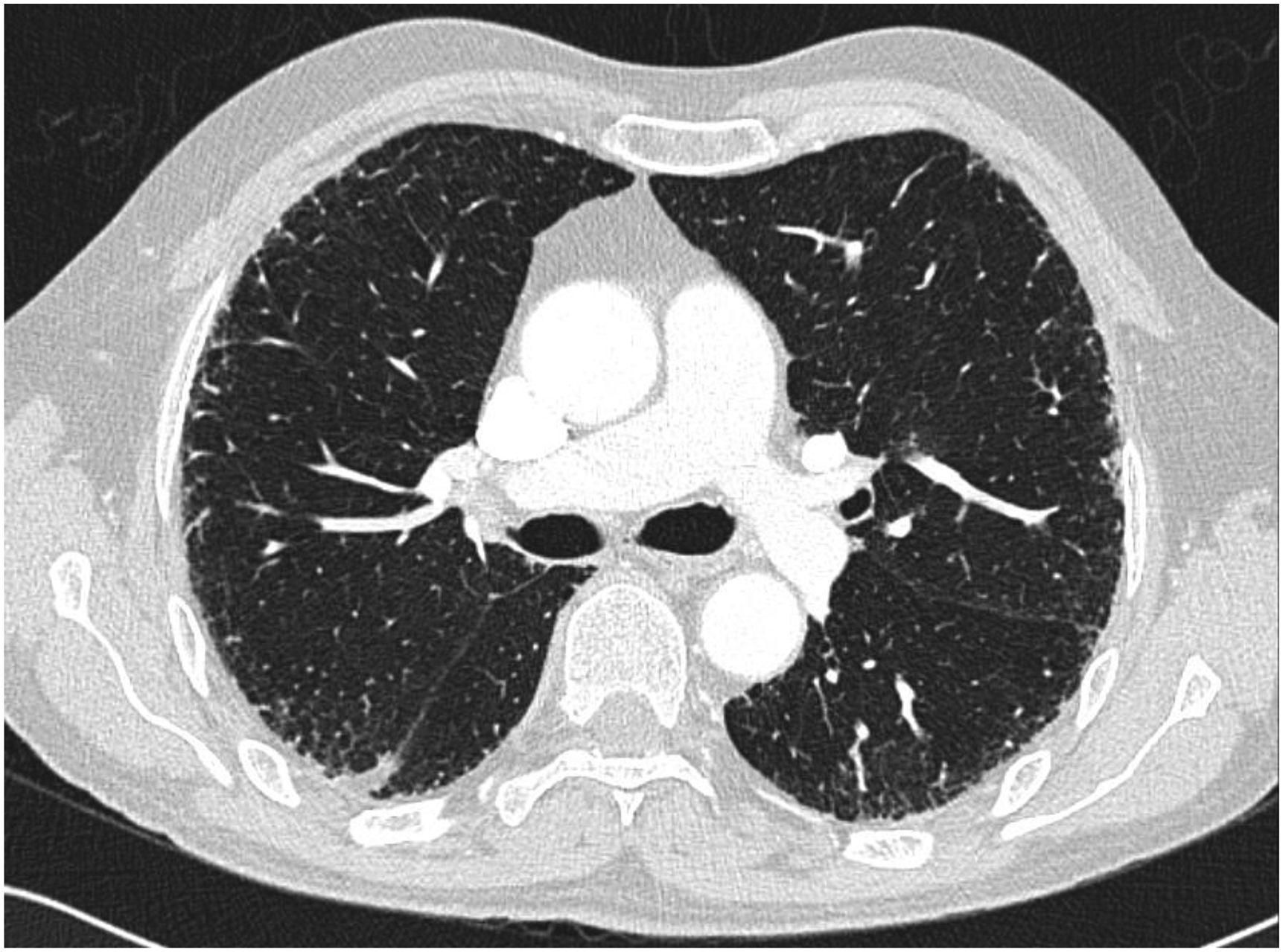
Dada la clínica actual y la afectación pulmonar previa no catalogada tras constatarse en la radiografía simple de tórax una afectación retículo intersticial bilateral, se completó el estudio con un TC torácico, objetivando un patrón de afectación pulmonar intersticial compatible con una neumonitis por hipersensibilidad versus una afectación tipo NINE. Además, destacaba una imagen nodular retroareolar derecha de 21 mm. Dada la posibilidad de un proceso neoplásico y una clínica de PNP paraneoplásica se completó el estudio mediante ecografía de senos y mamografía, observándose el tejido fibroglandular normal. En las imágenes abdominales se describe exclusivamente un nódulo prostático inespecífico (fig. 1).

En colaboración con el Servicio de Neurología y Neurofisiología de nuestro centro, se realizó un electromiograma, donde se objetivó una neurografía sensitiva del nervio sural y peroneo bilateral abolida, y motora ciático-poplíteo interno y externo no excitables. A nivel del tibial anterior derecho se observó fibrilación y un patrón de contracción neurógeno. Todo ello compatible con PNP sensitiva motora con degeneración axonal de intensidad grave.
Basado en los hallazgos clínicos y resultados de las exploraciones complementarias detallados previamente, se desarrolló un diagnóstico diferencial.
Diagnóstico diferencialEn resumen, se trata de un varón de 76 años, natural de Perú, que consultó por debilidad e hipoestesia de los miembros inferiores de una semana de evolución con un síndrome diarreico autolimitado. En los antecedentes se destacaba una PNP/mononeuritis múltiple diagnosticada en 2010 secundaria a una vasculitis sistémica p-ANCA (MPO) y una fibrosis pulmonar desde 2016. En la exploración física destacaba una disminución de la fuerza e hipoestesia infrarrotulianas. Dentro de las pruebas complementarias no mostraba alteraciones analíticas relevantes, unos anti MPO positivos y un FR fuertemente positivo, un sedimento urinario inactivo y un cuantiferón negativo. Se le realizó un electromiograma que informaba de una PNP sensitiva motora con degeneración axonal de intensidad severa más acusada en nervios inferiores. El estudio inicial con las pruebas de imagen mostraba un nódulo mamario BI-RADS 2 y una enfermedad intersticial pulmonar con zonas de panalización, bronquiectasias por tracción y áreas de atrapamiento aéreo. Se practicó una prueba que confirmó el diagnóstico.
Para iniciar el diagnóstico diferencial parece claro que el signo guía del cual partiremos será la PNP, ya que es la sintomatología más importante del paciente, así como el hallazgo más relevante en las pruebas complementarias. No obstante, disponemos de otros síntomas y signos que nos permitirán guiarnos en la resolución del caso y el eventual diagnóstico final.
El diagnóstico diferencial de las PNP puede ser muy amplio y complejo, ya sea por la variedad de enfermedades con la que pueden presentarse como por la diversidad de sus formas y su clasificación teniendo en cuenta el componente desmielinizante, axonal, sensitivo y/o motor. Así pues, es conveniente iniciar el diagnóstico diferencial con los 8 grandes grupos de situaciones clínicas o enfermedades que podrían desarrollar una PNP: enfermo crítico, oncológicas, metabólicas, infecciosas, genéticas, tóxicos, fármacos y enfermedades sistémicas. Dentro de todos estos, el que parece menos probable por el tiempo de evolución y las características del paciente, es el de enfermo crítico, razón por la cual lo descartamos ya desde un inicio.
Desde el punto de vista farmacológico, la lista de fármacos descritos que se han relacionado con una PNP principalmente axonal es muy extensa, siendo los más conocidos amiodarona, disulfiram, isoniazida, metronidazol, piridoxina, talidomida y quimioterápicos como taxanos y platinos. Si bien es verdad que en ocasiones la anamnesis farmacológica es complicada por el desconocimiento del paciente, en este caso, dados los antecedentes patológicos, no impresiona que esta sea la causa de la PNP1. Del mismo modo, tampoco aparece en la anamnesis una historia de consumo de tóxicos, ya sea voluntaria o involuntaria. Se han descrito casos de PNP relacionadas con productos como los organofosforados, acrilamida, arsénico, metales pesados o hexanos. En México, América Central y la zona norte del sur de América se han descrito intoxicaciones crónicas por la ingesta del fruto del arbusto conocido como tullidora o coyotillo (Karwinskia humboldtiana), presentando una PNP desmielinizante y una afectación renal2. Para poder descartarla, precisaríamos de una anamnesis muy dirigida. No obstante, por la rápida evolución del cuadro que nos ocupa y la ausencia de afectación renal, parece una opción muy poco probable.
Respecto a las enfermedades genéticas, las causas más comunes son las diferentes formas de Charcot-Mary-Tooth, si bien es verdad que principalmente son desmielinizantes y de afectación en edades mucho más tempranas que las del paciente. Dentro de ellas, la Charcot-Mary Tooht-2 se caracteriza por presentar daño axonal, aunque suelen presentarse antes de la quinta o sexta décadas de la vida, con importante atrofia muscular y con una clínica progresiva que el paciente no refiere. En este mismo grupo nos encontramos con las amiloidosis hereditarias, con PNP que debutan antes de los 70 años y con una marcada distribución geográfica, siendo Brasil y Bolivia los principales países afectos en el sur de América, sin casos descritos en Perú hasta la actualidad3. Otras causas de PNP hereditarias como las porfirias, ataxia de Friederich o ataxia telangiectasia parecen improbables con las características y la clínica descrita por el paciente.
Dentro de las causas oncohematológicas, están descritos los síndromes paraneoplásicos en forma de PNP, siendo principalmente axonales y sensitivas, con una rápida progresión. Dentro de las neoplasias de órgano sólido, los carcinomas son los más frecuentes, siendo el pulmonar el que lidera la lista, seguido de la próstata, la mama y las neoplasias endocrinas. Está descrita la presencia del anticuerpo anti-Hu en las formas paraneoplásicas subagudas o progresivas. Desde el punto de vista hematológico, tanto linfomas, mieloma múltiple y policitemia vera pueden presentar una PNP como síndrome paraneoplásico, en muchas ocasiones precediendo la neoplasia4.
En cuanto a las causas metabólicas, si bien son muchas las enfermedades que pueden presentar algún tipo de afectación neuropática, las más frecuentes son el trastorno por abuso de alcohol o la neuropatía asociada a diabetes mellitus, ambas etiologías descartadas desde un principio con la anamnesis inicial. Otras causas como el déficit de vitamina B12, el hipotiroidismo u otros trastornos hormonales no parecen jugar un papel en el caso que nos ocupa.
Si nos adentramos en las enfermedades infecciosas, la infección por el VIH se presenta como una de las etiologías más ampliamente conocidas. No obstante, se presenta en forma de PNP axonal, principalmente sensitiva y de curso crónico. Si tenemos en cuenta que las serologías durante el ingreso fueron negativas, esta etiología quedaría descartada. La enfermedad de Lyme puede presentarse como una PNP subaguda o crónica, con afectación radicular y en ocasiones de pares craneales. El 50-80% de los casos presentan el característico eritema migratorio y dolor osteoarticular generalizado, signos y síntomas que no se hallaron en este caso. Un caso particular son las infecciones relacionadas con el síndrome de Guillain-Barré (SGB), en concreto el virus del Zika si tenemos en cuenta el origen del paciente (Perú). En 2018, cuando el paciente ingresó, se habían descrito varios casos de SGB asociados a la infección por el virus del Zika en Colombia y Perú 5.
Para finalizar, nos centraremos en las enfermedades sistémicas. Las enfermedades sistémicas son un grupo de enfermedades que como su nombre indica, se pueden presentar afectando a cualquier órgano y con una variedad de signos y síntomas que las hace difíciles de agrupar. No obstante, trataremos de centrarnos en 4 grandes grupos que podrían presentar una PNP, como son la sarcoidosis, la mielitis transversa, las enfermedades del tejido conectivo y el SGB. La sarcoidosis es una enfermedad granulomatosa con una gran variedad de formas de presentación, motivo por el cual se la conoce como la gran simuladora. Si bien es verdad que en Perú es una enfermedad poco frecuente y, más aún, su forma de presentación únicamente como PNP, la biopsia en este caso de algún nervio periférico sería la que confirmaría o descartaría el diagnóstico. Focalizándonos en las enfermedades del tejido conectivo, se debe hacer el diagnóstico diferencial con aquellas entidades que puedan presentarse como una neuropatía vasculítica, como las vasculitis ANCA, poliarteritis nodosa (PAN), crioglobulinemia y neuropatía vasculítica no sistémica. Dentro de estas, las vasculitis ANCA podrían parecer las más razonables. La poliangeítis microscópica (PAM) es una vasculitis de pequeño vaso, con anticuerpos ANCA especificidad MPO positivos y que afecta a distintos órganos en forma de púrpura, glomerulonefritis, mononeuritis múltiple, hemorragia alveolar y hasta fibrosis pulmonar. El caso que nos ocupa presenta ANCA-MPO positivos, fibrosis pulmonar y afectación neuropática, si bien es verdad que no parece una mononeuritis múltiple. Asimismo, no presenta afectación de otros órganos diana que nos ayudaría a focalizar el diagnóstico. No obstante, una vez más la biopsia nos daría el diagnóstico. La otra entidad de la que podría tratarse este caso sería la neuropatía vasculítica no sistémica, con la presencia de daño axonal en múltiples nervios de forma individual, asimétrica y la necesidad de una biopsia de nervio para el diagnóstico. Sobre la mielitis transversa, es una entidad en la que actualmente se habla más del espectro de las mielitis transversas, englobando desde causas infecciosas y paraneoplásicas hasta las diferentes formas de las neuromielitis ópticas. Son entidades que suelen tener afectación parcheada, asociando tanto síntomas motores, sensitivos como autonómicos y con la afectación de primera motoneurona. El diagnóstico suele precisar de resonancia medular y del sistema nervioso central, así como de análisis del líquido cefalorraquídeo6,7. Finalmente nos focalizaremos en el SGB. El SGB es una enfermedad considerada autoinmunitaria que se caracteriza por una debilidad simétrica, rápidamente progresiva, de afectación inicialmente distal y después ascendente hasta llegar a afectar la musculatura respiratoria en algunos casos, así como afectación disautonómica hasta en el 70% de los casos. Como desencadenantes se conocen gran cantidad de infecciones víricas y bacterianas, de entre las más conocidas la infección por Campylobacter, CMV, VEB, Zika, VIH y enterovirus. No obstante, se han descrito otros desencadenantes como vacunas, cirugías, traumatismos e incluso trasplante de progenitores hematopoyéticos. La forma aguda suele presentarse con una evolución de 3 a 7 días, aunque existen varios subtipos como la desmielinizante aguda inflamatoria (AIDP), la axonal aguda sensitivo-motora (AMSAN) o motora pura (AMAN) y la debilidad parafaríngea-cervico-braquial, con anticuerpos antigangliósidos (GM1, GD1 o GQ1B) como datos diferenciales de estas entidades8. Las formas crónicas ya se clasificarían como otra entidad por separado del SGB, como serían la PNP desmielinizante idiopática crónica (CIDP) o su variante axonal (CIAP), con un curso clínico superior a 8 semanas9. Para el diagnóstico del SGB, se deberían cumplir los criterios de Brighton10, siendo necesarias una clínica y evolución compatible, un EMG congruente y un análisis del líquido cefalorraquídeo con disociación albúmino-citológica.
Resumiendo, los diagnósticos que nos planteamos son la posibilidad de una vasculitis, un SGB variante axonal (AMSAN) o un síndrome paraneoplásico. Para la primera, los ANCA-MPO, la fibrosis pulmonar y la elevación de factor reumatoide respaldarían el resultado, precisando de una biopsia del nervio sural para su confirmación. Por otro lado, el paciente presenta una PNP sensitivo-motora y un cuadro diarréico, haciendo posible el SGB. Para proseguir en el diagnóstico precisaríamos de una punción lumbar, así como unos anticuerpos antigangliósidos que ayudarían al diagnóstico. Finalmente se podría tratar de un síndrome paraneoplásico dado que el paciente presentaba un nódulo prostático no descrito previamente, así como parte de una neoplasia oculta, precisando en este caso un estudio con PSA y eventual PET-TC. En conclusión, y con todos los datos expuestos, nuestra primera opción sería la de un SGB variante axonal, solicitando un análisis del líquido cefalorraquídeo y unos anticuerpos antigangliósidos.
Diagnóstico clínicoSDG variante AMSAN.
DiscusiónFinalmente, el Servicio de Traumatología de nuestro centro realizó una biopsia del nervio sural izquierdo que fue valorado de forma detallada por la Unidad de Anatomía Patológica, hallando en los fragmentos de tejido analizados fenómenos de desmielinización, remielinización, con degeneración y regeneración axonal. Así mismo, estructuras en capa de cebolla (fig. 2). Dado la afectación recurrente, la clínica, los hallazgos electromiográficos y la anatomía patológica compatible, el diagnóstico definitivo fue de PNP inflamatoria desmielinizante crónica (CIDP).

Clínicamente existen diferentes subtipos clínicos de CIDP en relación con el tipo y la localización de la afectación nerviosa. El subtipo clínico típico o más prevalente (entorno al 50-60%)11 se basa en la afectación simétrica sensitivo-motora distal y proximal, en el que generalmente se objetiva una progresión gradual de los síntomas durante 8 semanas. Sin embargo, en un pequeño porcentaje de pacientes se objetiva una progresión más aguda como sucede en nuestro caso clínico12. Como síntomas destacan la debilidad, la dificultad para la deambulación y la realización de trabajos motores. La afectación sensitiva se puede representar en forma de ataxia, dolor o parestesias y/o disminución o ausencia de reflejos13.
Ante una sospecha clínica de CIDP se debe iniciar el proceso diagnóstico y diferencial, como el citado previamente. Paralelamente existen exploraciones complementarias que pueden reforzar el diagnóstico. Entre ellas destaca el electromiograma (EMG) que estudia la funcionalidad sensitiva y motora nerviosa. Los hallazgos electrofisiológicos característicos son: decremento de la velocidad de conducción o aparición de bloqueos de conducción objetivados mediante el aumento de las latencias motoras distales y/o desaparición o retraso de las ondas F14. Sin embargo, estas alteraciones de función de conducción se pueden detectar en otras enfermedades que asocian degeneración (neuropatías, diabetes mellitus), razón por la cual es importante descartarlas en el proceso diagnóstico15. El electromiograma de nuestro paciente se objetiva una abolición de los potenciales y afectación axonal que también se ha descrito en la bibliografía médica como posible afectación nerviosa en los pacientes con CIDP16. Otra de las exploraciones recomendadas es el estudio del líquido cefalorraquídeo mediante una punción lumbar, ya que un alto porcentaje de los pacientes (alrededor de 80%) presenta disociación albuminocitológica, con una proteinoraquia > 45 mg/dl14. Se puede plantear la realización de pruebas de imagen para observar la morfología nerviosa, concretamente se utilizan en la práctica clínica la resonancia magnética con gadolinio y la ecografía de nervios, en los que se puede llegar a visualizar hipertrofia nerviosa17.
Inicialmente existía alta sospecha de vasculitis ANCA, además de positividad de los anticuerpos. Sin embargo, en algunas series de pacientes diagnosticados de CIDP se ha detectado positividad de los mismos (ANCA, ANA y factor reumatoide)18. En la bibliografía médica se recomienda que cuando existe una sospecha clínica de una posible enfermedad infiltrativa o vasculitis, que la exploración de elección sea una biopsia del nervio19. Dada la afectación parcheada nerviosa, la sensibilidad y especificidad de la prueba es baja, para aumentar la rentabilidad se escoge el nervio con mayor afectación clínica, como en nuestro caso el sural izquierdo.
La anatomía patológica compatible con CIDP muestra un edema endoneural, macrófagos asociados a las fibras nerviosas con procesos de desmielinización y remielinización, formaciones en «capa de cebolla», e infiltración celular endoneural14, hallazgos similares a los que se encontraron en la biopsia del nervio sural de nuestro paciente. También permitió descartar otras neuropatías, ya que no se objetivaron depósitos de rojo congo, granulomas, o afectación vascular.
La clínica inicial tanto en el SGB citado como probabilidad diagnóstica más alta como en la CIDP son muy similares, sin embargo, dada la recurrencia temporal y los hallazgos anatomopatológicos, nos inclinamos más por la sospecha de CIDP.
Consideramos este el diagnóstico final de nuestro paciente basándonos en los criterios de la Federación Europea de las Sociedades de Neurología y de Nervio Periférico, donde se define que el diagnóstico definitivo de CIDP se establece con base en una afectación clínica y electrofisiológica compatible junto con la suma de alguna de las exploraciones de imagen, anatomía patológica o líquido cefalorraquídeo, o la respuesta al tratamiento14.
Paralelamente en este caso clínico el paciente presentaba fibrosis pulmonar. En la bibliografía médica no se describe una asociación clínica entre ambas enfermedades, a excepción de un caso clínico publicado que describe la convivencia de ambas afectaciones20.
Al tratarse de una enfermedad autoinmune, el tratamiento se basa en la utilización de técnicas inmunomoduladoras. Los regímenes propuestos como tratamiento inicial son los glucocorticoides y las inmunoglobulinas. Como alternativa a los tratamientos previos se puede realizar plasmaféresis, aunque se considera un tratamiento menos efectivo y accesible por la técnica y el proceso invasivo21. En el caso clínico que nos ocupa tras la realización de la biopsia y a la espera de los resultados anatomopatológicos por la sospecha clínica, se administraron pulsos de metilprednisolona, seguidos de prednisona 40 mg al día, con una mejoría parcial de la sintomatología. Dada la extensión y la gravedad de la afectación neurológica se completó el tratamiento con 5 ciclos de inmunoglobulinas. Tras ello, el paciente presentó una recuperación prácticamente completa de la clínica neurológica, y mejoría de la conducción nerviosa en la electromiografía de control.
Los autores consideramos este caso clínico interesante, en primer lugar, por el amplio diagnóstico diferencial que se establece, además de las dudas diagnósticas que plantean los propios resultados de las exploraciones solicitadas. La positividad de los anticuerpos puede orientar al clínico hacia un rápido diagnóstico de vasculitis, como sucedió en el primer episodio clínico que sufrió nuestro paciente en su país de origen. La afectación pulmonar intersticial podría actuar como un factor distractor, ya que se encuentra más relacionada con una posible vasculitis de pequeño vaso. Sin embargo, en la situación clínica de este paciente como opción más plausible, se podría tratar de una neumopatía por exposición laboral o ambiental dado que la coexistencia de CIDP y la afectación intersticial es un mero caso anecdótico en la bibliografía médica.
Consideramos que en casos clínicos similares, ante la duda diagnóstica se podría optar por la realización de la biopsia de nervio periférico, aunque no sea la prueba diagnóstica más sensible u específica para la CIDP, dado que da soporte en el diagnóstico diferencial. Finalmente, puesto que el paciente regresó a su país de origen, no se pudo continuar el seguimiento clínico y evolutivo posterior que podría ayudar a la mejor caracterización de la enfermedad de base, la afectación intersticial pulmonar o a los hallazgos durante el diagnóstico diferencial como el nódulo mamario y prostático.
DiagnósticoCIDP.
Responsabilidades éticasSe han seguido los procedimientos éticos del centro para la publicación de este artículo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
