




La arteritis de células gigantes es una vasculitis sistémica con importante afectación intra y extracraneal que, con diagnóstico precoz y tratamiento en Atención Primaria, puede mejorar su pronóstico al tratarse de una emergencia médica.
Nuestro grupo de trabajo de vasculopatías de la Sociedad Española de Médicos de Atención Primaria (SEMERGEN) ha elaborado una serie de recomendaciones con la evidencia científica actual para su abordaje y seguimiento de una manera multidisciplinar desde atención primaria.
Giant cell arteritis is a systemic vasculitis with significant intra and extracranial involvement that, with early diagnosis and treatment in primary care, can improve its prognosis as it is a medical emergency.
Our working group on vascular diseases of the Spanish Society of Primary Care Physicians (SEMERGEN) proposes a series of recommendations based on current scientific evidence for a multidisciplinary approach and follow-up in primary care.
La arteritis de células gigantes (ACG) también conocida como enfermedad de Horton, arteritis craneal o arteritis de la temporal, es una vasculitis inflamatoria granulomatosa no necrotizante, que afecta a las arterias de mediano y gran calibre. Las arterias carótidas externas e internas y sus ramas, son las que con mayor frecuencia se ven afectadas, responsables de las manifestaciones clínicas clásicas de la enfermedad1: cefalea, claudicación mandibular y alteraciones visuales. A menudo, coexiste la afectación extracraneal de grandes vasos (ACG-GV) fundamentalmente en la región del cayado aórtico y de la aorta torácica.
Se puede considerar, por tanto, que el espectro clínico de ACG incluye dos subtipos con distintos mecanismos patológicos, la ACG clásica con afectación predominante de ramas craneales aisladas y la de la ACG-GV2.
Las manifestaciones y complicaciones de la ACG pueden llevar a una importante morbimortalidad si no se diagnostican y tratan precozmente por lo que es necesario conocer y valorar de forma adecuada esta emergencia médica desde Atención Primaria.
EpidemiologíaLa ACG es una vasculitis que afecta predominantemente a individuos de raza caucásica, mayores de 50 años. En la mayoría de las series estudiadas, la edad media es de 70 años o superior, aumentando su incidencia con la edad, de tal forma que es casi cinco veces más frecuente en la novena década de la vida. La incidencia anual estimada de ACG oscila entre el 1,6 a 32,8/100.000 individuos mayores de 55 años de edad, afecta más a mujeres que a hombres, en una proporción de 2-4:13–5, varía según el área geográfica, siendo más frecuente en los países escandinavos y en comunidades con ascendencia escandinava como el condado de Olmsted, Minnesota, donde la población tiene un fuerte origen escandinavo lo que apunta a la importancia de factores etiológicos que varían latitudinalmente, siendo superiores a 20/100.000. En el sur de Europa e Israel, por el contrario, la incidencia es inferior a 12/100.000 en mayores de 50 años. La ACG es casi excepcional en la población negra, en Japón y en el norte de la India6.
En España, un estudio epidemiológico realizado en Lugo entre 1981 a 2005 mostró una tasa de incidencia anual ajustada por edad y sexo de ACG confirmada mediante una histología positiva de la biopsia de la arteria temporal de 10,13 por 100.000 habitantes en mayores de 50 años. La tasa de incidencia anual en mujeres fue ligeramente mayor. La incidencia anual va aumentando con la edad hasta un máximo de 23,16 por 100.000 habitantes en el grupo de edad de 70 a 79 años7.
La supervivencia no parece afectarse por la aparición de la enfermedad, presentando los pacientes con ACG una esperanza de vida similar a su población control por edad y sexo8,9.
La epidemiología de la ACG se ha centrado históricamente en los pacientes con síntomas predominantemente craneales, debido fundamentalmente a los criterios de clasificación de The American College of Rheumatology (1990)10–11.
EtiopatogeniaLa etiología sigue siendo desconocida, si bien, la alta prevalencia de ACG entre europeos del norte y estadounidense con ascendencia étnica similar, aboga hacia una predisposición hereditaria de la enfermedad, habiéndose demostrado una asociación genética significativa al antígeno leucocítico humano (HLA)-DR4 y un polimorfismo de secuencia expreso codificado en el interior de la región hipervariable del gen HLA-DRβ1*04. Sobre esta predisposición genética actuarían factores ambientales e infecciosos. La existencia de factores ambientales lo indicarían los agrupamientos geográficos de los casos, así como la asociación entre el tabaquismo y el riesgo aumentado en las mujeres de presentar la enfermedad, que ha sido constatado en diferentes estudios12,13. Las hipótesis formuladas por algunos autores sobre una posible relación con agentes infecciosos como el virus de la varicela zoster, el mycoplasma o la Chlamydia pneumoniae, el parvovirus B19 y el virus paragripal de tipo II, tras detectar en biopsias de arterias temporales, moléculas virales o relacionar incidencias con brotes infecciosos, hasta ahora no se han podido demostrar14–17.
Los cambios histológicos clásicos de la ACG son consecuencia de la inflamación vascular con infiltración de linfocitos, macrófagos, células gigantes y de la formación de granulomas, que conducen a una hiperplasia de la íntima y de una oclusión de la luz arterial con destrucción de la pared del vaso18. El término arteritis granulomatosa hace referencia a ese hallazgo anatomopatológico, aunque hay que tener en cuenta que las células gigantes multinucleadas faltan aproximadamente en la mitad de los casos biopsiados y pueden estar presentes en otras formas de vasculitis, por lo que su hallazgo no es necesario para formular el diagnóstico si otras características son compatibles.
Actualmente, la fisiopatología de la ACG se puede esquematizar en dos ejes que explican los síntomas clínicos, la respuesta inflamatoria sistémica y la oclusión vascular, existiendo una interacción compleja entre los sistemas inmunitarios innato y adaptativo19–21. Del sistema inmunológico innato depende la respuesta inflamatoria sistémica, de tal forma que las células de este sistema inmunológico (células dendríticas vasculares y monocitos) producen citocinas proinflamatorias como la interleucina 6 (IL-6) que están asociadas con la producción de proteínas de fase aguda en el hígado (principalmente proteína C reactiva). La respuesta inflamatoria sistémica es sensible a glucocorticoides (GC) y a anti-IL-6.
La oclusión vascular debida al remodelado vascular, da como resultado la complicación isquémica. La activación de los macrófagos o las células del músculo liso vascular lesionadas producen factores de crecimiento, derivados de plaquetas y de tejido endotelial vascular, que desencadenan la remodelación y una diferenciación de miofibroblastos de las células del músculo liso del vaso, produciendo hiperplasia de la íntima y oclusión luminal, que tiene como consecuencia las complicaciones clínicas (ceguera, accidente cerebrovascular y síndrome del arco aórtico) de la ACG. Esta remodelación vascular no se ve afectada por los GC y la terapia anti-IL-621.
Manifestaciones clínicasLas manifestaciones clínicas de la ACG suelen ser muy variadas e inespecíficas al inicio, generalmente se presentan de forma silente, aunque en ocasiones los síntomas pueden aparecer de forma abrupta22,23, estas son causadas por la inflamación arterial (principalmente arterias carótidas y sus ramas) o pueden formar parte de un cuadro constitucional con fiebre de origen desconocido.
La cefalea es la manifestación característica, sobre todo en las formas clásicas craneales y la describen alrededor de un 70 a 80% de estos pacientes al inicio de la enfermedad, presentando anomalías en la exploración de las arterias temporales que puede ser claramente visible como un cordón prominente, indurado, tortuoso, sensible y doloroso a la palpación. Puede ser hemicraneal, temporal o bitemporal, en algunos casos puede ser holocraneal y raramente occipital. La afectación de otras ramas craneales derivadas de la carótida externa puede ser causa de claudicación mandibular, dolor facial o, incluso, de necrosis lingual. El paciente con ACG predominantemente craneal, a menudo experimenta claudicación a la masticación, sobre todo para alimentos sólidos. Puede incluso referir disfagia o dolor referido en encías al masticar.
La manifestación más temida es el desarrollo de pérdida de visión que en la mayoría de los casos, es inicialmente unilateral y aparece antes del inicio de la corticoterapia o a veces, poco tiempo después, siendo excepcional su aparición en las recaídas24,25. La pérdida de visión puede ser total (amaurosis) o consistir en reducciones del campo visual (hemianopsias o cuadrantanopsias). En más del 50% de los casos, la afectación visual definitiva está precedida de pródromos transitorios: visión borrosa, diplopia, fosfenos o amaurosis fugaz.
La afectación vasculítica de las arterias ciliares posteriores produce, además, una neuropatía óptica isquémica.
Desde que se utiliza sistemáticamente la corticoterapia, la frecuencia de complicaciones visuales se ha reducido a la mitad, siendo del 25 al 30%; con una ceguera irreversible, en el 10 a 18% de los casos, aunque estas cifras varían según los estudios24,25.
Se ha descrito, como consecuencia de la afectación vasculítica en el territorio vertebro-basilar, el desarrollo de infartos cerebrales en un 3 a 6% de los pacientes, produciendo cegueras corticales26.
No es raro que, en el momento del diagnóstico, los pacientes presenten hipoacusia y vértigos debido a disfunción vestibular. Es importante notar que la clínica vestibular a menudo se recupera con el tratamiento esteroide27.
Por otro lado, la afectación vasculítica de las arteriales extracraneales presentan unas manifestaciones clínicas diferentes, como son la insuficiencia cardiaca, el dolor torácico o la muerte súbita, debidos a la formación de aneurismas, disección y rotura de las arterias. En personas con ACG es prudente realizar un control periódico, al menos anual, vigilando los factores que predisponen al desarrollo de aneurismas, sobre todo la hipertensión arterial, más comunes en la aorta torácica28. Suelen ocurrir generalmente en el seguimiento de los pacientes, años después del diagnóstico de la ACG y no al inicio de la enfermedad.
En un 10 a 15% de pacientes se presentan síntomas derivados de la estenosis de los grandes vasos, fundamentalmente claudicación intermitente de extremidades29.
La afección de las arterias coronarias y mesentéricas es poco frecuente. Encontramos manifestaciones sistémicas como astenia, anorexia, pérdida de peso y fiebre o febrícula, aunque con menor frecuencia.
Merece especial atención la polimialgia reumática (PMR) que, si bien este síndrome no es una manifestación misma de la enfermedad, está presente en aproximadamente la mitad de los pacientes con ACG. Cursa con dolor en las cinturas escapular y pelviana que se exacerba con la movilización y rigidez, especialmente matutina.
La presencia de una PMR asociada a la ACG es más común en las formas extracraneales y puede ser la primera manifestación de la ACG. Con frecuencia, pacientes con PMR aparentemente aislada muestran afectación de grandes vasos cuando se realiza una prueba de imagen, como la tomografía axial por emisión de positrones (PET-TC)30–33.
Criterios diagnósticosLos criterios de clasificación clásicos para la ACG fueron descritos en 1990 por la American College of Rheumatology. La presencia de tres o más criterios se considera como asociada a alta posibilidad diagnóstica de ACG:
- -
Edad mayor a 50 años al inicio de los síntomas.
- -
Cefalea de reciente aparición o distinta a la existente (con sensibilidad en cuero cabelludo).
- -
Anomalías en la arteria temporal (dolor o hipersensibilidad a la palpación, disminución o ausencia de pulso)
- -
Velocidad de sedimentación globular (VSG) > 50 mm/h en la primera hora.
- -
Biopsia de la arteria temporal que demuestre vasculitis con predominio de infiltrado mononuclear y formación de granulomas.
El diagnóstico de ACG en Atención Primaria sigue siendo difícil debido a la naturaleza inespecífica de muchos de sus síntomas tempranos similares a otras patologías, siendo una enfermedad infrecuente lo que dificulta que se pueda considerar dentro del diagnóstico diferencial en primera instancia, por ello, el retraso en el diagnóstico no es inusual. La importancia de comprender el alcance del retraso en el diagnóstico y las razones asociadas con el mismo han sido ampliamente investigadas para mejorar la atención a estos pacientes. Prior et al.34 examinaron el tiempo de retraso entre la primera aparición de síntomas relacionados con la ACG y el diagnóstico de confirmación de la misma, encontrando que el retraso medio era de nueve semanas y que la demora, incluso cuando los pacientes presentaban síntomas craneales, era de ocho semanas la cual se alargaba hasta 18 en aquellos con síntomas no craneales. En vista de las consecuencias potencialmente graves de un retraso en el diagnóstico de ACG, dado que la probabilidad de recuperación visual es baja una vez que se ha establecido la ceguera, el reducir este retraso sería evidentemente beneficioso y podría resultar en una disminución de costes para el sistema sanitario35–38.
Pruebas complementariasSe pueden utilizar algunos parámetros clínicos y de imagen:
-Reactantes de fase aguda: la VSG es el biomarcador fundamental para el diagnóstico y monitorización de la ACG. Una VSG mayor a 50 mm/h constituye uno de los criterios diagnósticos, no obstante, un valor de VSG normal no excluye la posibilidad del diagnóstico, ya que se ha descrito que hasta un 20% de pacientes con ACG pueden presentar una VSG menor a 50 mm/h22.
Un estudio que incluyó una serie de 240 personas con ACG clásica confirmada por biopsia mostró valores de hemoglobina inferiores a 12 g/dL, alrededor del 55% de los casos, trombocitosis en casi la mitad de los pacientes y presencia de marcadores de inflamación como la elevación de fosfatasa alcalina, leucocitos, proteína C reactiva, VSG e hipoalbuminemia. La anemia se observó con mayor frecuencia en pacientes sin manifestaciones isquémicas graves y en aquellos con síndrome constitucional o fiebre39.
Confirmando que existe mayor riesgo de complicaciones oculares graves en aquellos individuos con ACG que presentan una VSG entre 70 y 100 mm/h, mientras que con VSG mayor de 100 mm/1ª hora suelen tener menor incidencia de eventos isquémicos visuales40.
Métodos de diagnóstico-Biopsia de la arteria temporal: considerada el patrón oro para el diagnóstico de la ACG. Una biopsia positiva confirma el diagnóstico, mientras que una negativa no puede descartarlo, ya que la inflamación tiende a afectar las arterias de forma segmentaria o parcheada, y esto hace que en ocasiones la biopsia pueda no ser diagnóstica si no se realiza eco-guiada.
Un resultado normal en la biopsia bilateral tiene un valor predictivo negativo del 90%. El rendimiento de la biopsia de la arteria temporal es alto en pacientes con ACG craneal clásica. Sin embargo, es mucho menor y generalmente no superior a un 40% en personas con ACG de predominio extracraneal31.
-Ecografía Doppler (eco-D): los primeros estudios con eco-D en sujetos con ACG sugirieron que el signo del halo (un área oscura hipoecogénica alrededor de la pared del vaso debido a edema de la pared vascular) era muy específico de ACG, demostrando una sensibilidad del 77% y una especificidad del 96%. Además de ser de utilidad para guiar la biopsia de la arteria temporal, la presencia de este signo tiene una especificidad del 100% para el diagnóstico de la enfermedad comparado con la biopsia. Además, se sabe que estos hallazgos ecográficos desaparecen tras iniciar el tratamiento, por lo que esta técnica podría ser incluso útil en el seguimiento de estos pacientes.
En personas en las que se sospeche una ACG, un test de imagen temprano como puede ser la eco-D está recomendado por la European League Against Rheumatism (EULAR)41,42 como criterio a tener en cuenta para el diagnóstico, si bien es verdad que la realización de la prueba de imagen no debe retrasar el inicio del tratamiento. En aquellos casos en los que la sospecha clínica sea elevada y el resultado de la ecografía sea positivo, el diagnóstico de ACG puede establecerse sin necesidad de pruebas complementarias adicionales.
Por otro lado, ha sido también descrita la utilidad de la ecografía en el estudio de las arterias extracraneales que pueden estar afectadas en la ACG.
Todo esto, junto con fácil disponibilidad de la técnica en algunos de los centros de salud en España y lo barato del procedimiento hace que esta prueba sea recomendada por la EULAR como la primera prueba de imagen indicada en pacientes con sospecha predominante de ACG. Algunos autores defienden la inclusión de los hallazgos ecográficos típicos en los criterios diagnósticos de la enfermedad22,43.-Otras pruebas: ante la sospecha de afectación de grandes vasos extracraneales se solicitará tomografía axial computarizada (TAC) o resonancia magnética nuclear para valorar la aorta y sus principales ramas, ya que se ha demostrado que un 54% de los individuos diagnosticados de ACG y/o PMR presentan inflamación subclínica de la aorta o sus ramas. La tomografía axial por emisión de positrones (PET/TAC) puede ser útil en pacientes no respondedores al tratamiento con GC o en manifestaciones sistémicas atípicas, permitiendo realizar un diagnóstico precoz de ACG en formas extracraneales44–46.
Diagnóstico diferencialDeberemos descartar una vasculitis como causa de la enfermedad en aquellos pacientes con cefalea, sobre todo en aquellos que sean de edad avanzada, con cefalea de reciente comienzo o distinta de la habitual y/o que presentan manifestaciones craneales con/sin síndrome constitucional. El diagnóstico de vasculitis será apoyado por la elevación de los biomarcadores y los hallazgos específicos en las pruebas de imagen.
Las patologías que pueden presentarse de forma similar a ACG serían las siguientes:
- •
Enfermedades virales, tuberculosis o endocarditis infecciosa: pueden presentar poliartralgias en el contexto de la fiebre.
- •
Neoplasias sólidas o hematológicas: por la presencia de síntomas constitucionales y dolor muscular proximal.
- •
Bursitis subacromial o artritis reumatoide que pueden afectar las articulaciones proximales, aunque la artritis reumatoide es más típica de pequeñas articulaciones.
- •
Dermatomiositis y polimiositis.
- •
Migraña
- •
Hipotiroidismo ya que puede provocar dolor, rigidez y artralgias. De manera más específica, se puede objetivar en la exploración un enlentecimiento de los reflejos tendinosos profundos23.
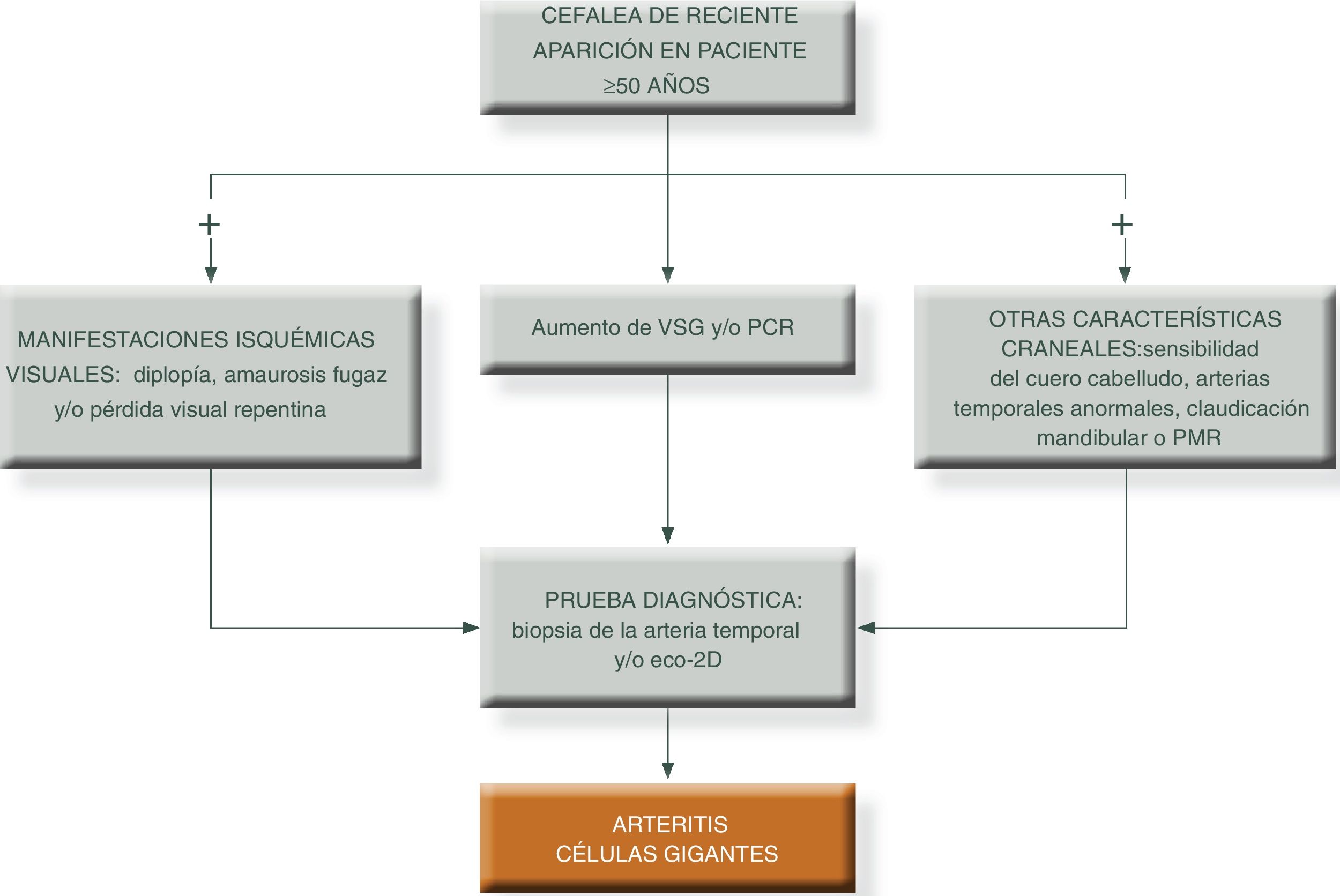
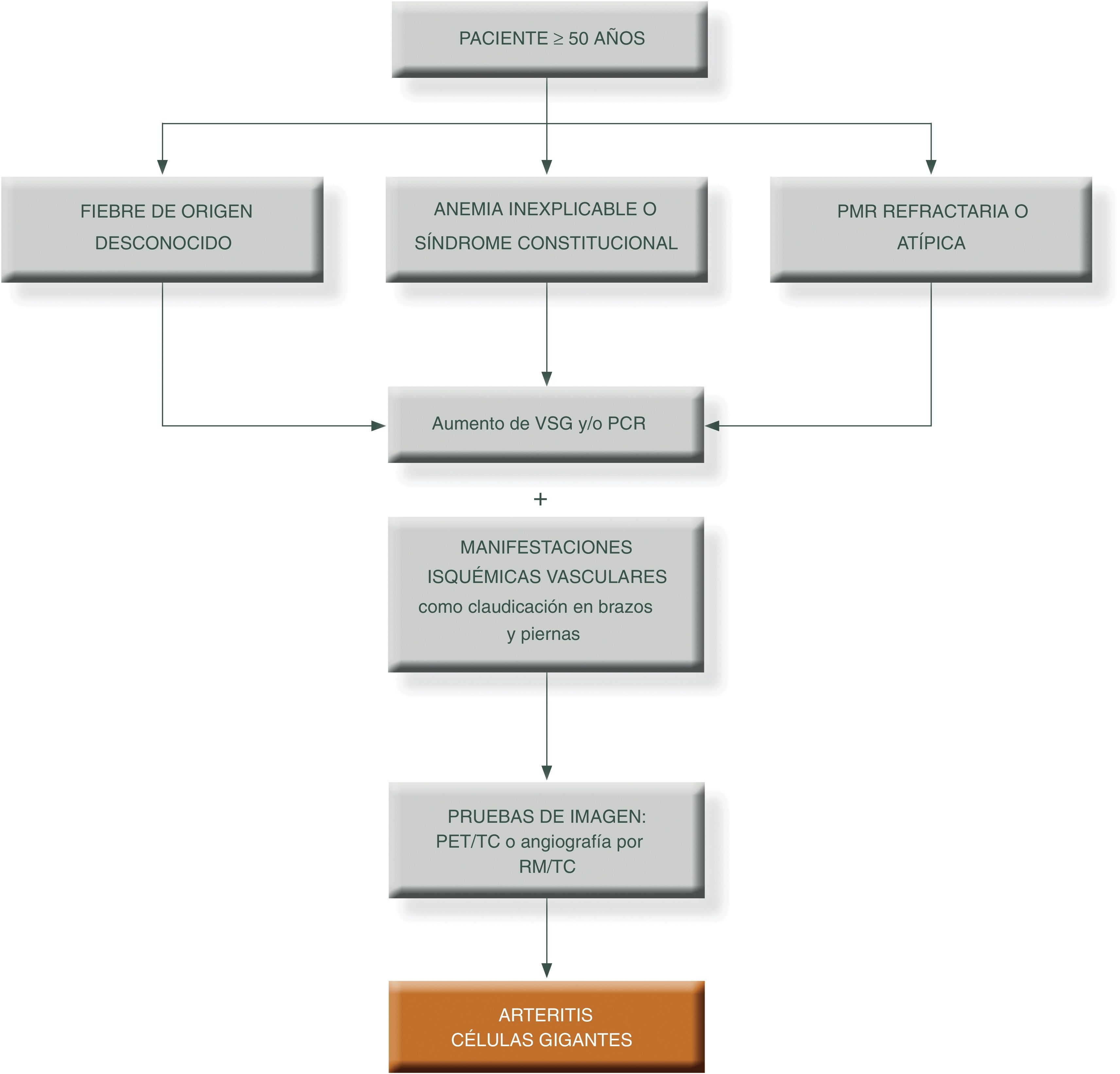
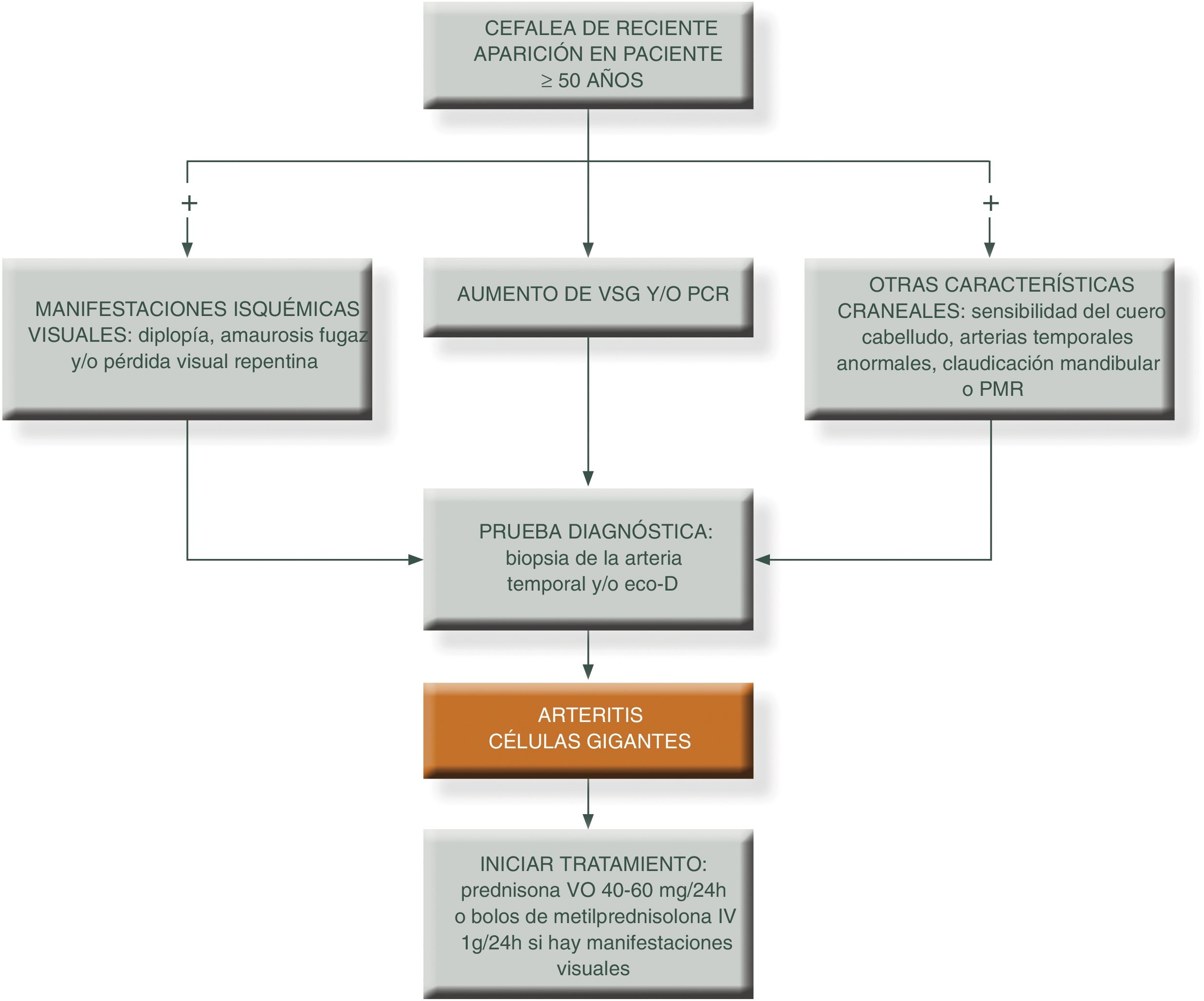
Para simplificar el manejo de estos pacientes y aumentar el grado de sospecha clínica, podemos presentar los siguientes algoritmos descritos en las figuras 1 y 231.


Algoritmo diagnóstico en pacientes con patrón extracraneal típico de vasculitis de grandes vasos.
PCR: proteína c reactiva; PET/TAC: tomografía axial por emisión de positrones; PMR: polimialgia reumática; RM/TC: resonancia magnética nuclear; TC: tomografía computarizada; VSG: velocidad de sedimentación globular.
Fuente: elaboración propia.
El objetivo del tratamiento es mejorar los síntomas y evitar el riesgo de complicaciones isquémicas graves, sobre todo el desarrollo de ceguera, en particular en personas que se presentan con la forma clásica craneal. En este sentido, si el paciente ya ha sufrido pérdida de visón de un ojo debemos instaurar rápidamente tratamiento con GC a dosis altas para tratar de evitar el desarrollo de ceguera en el ojo contralateral.
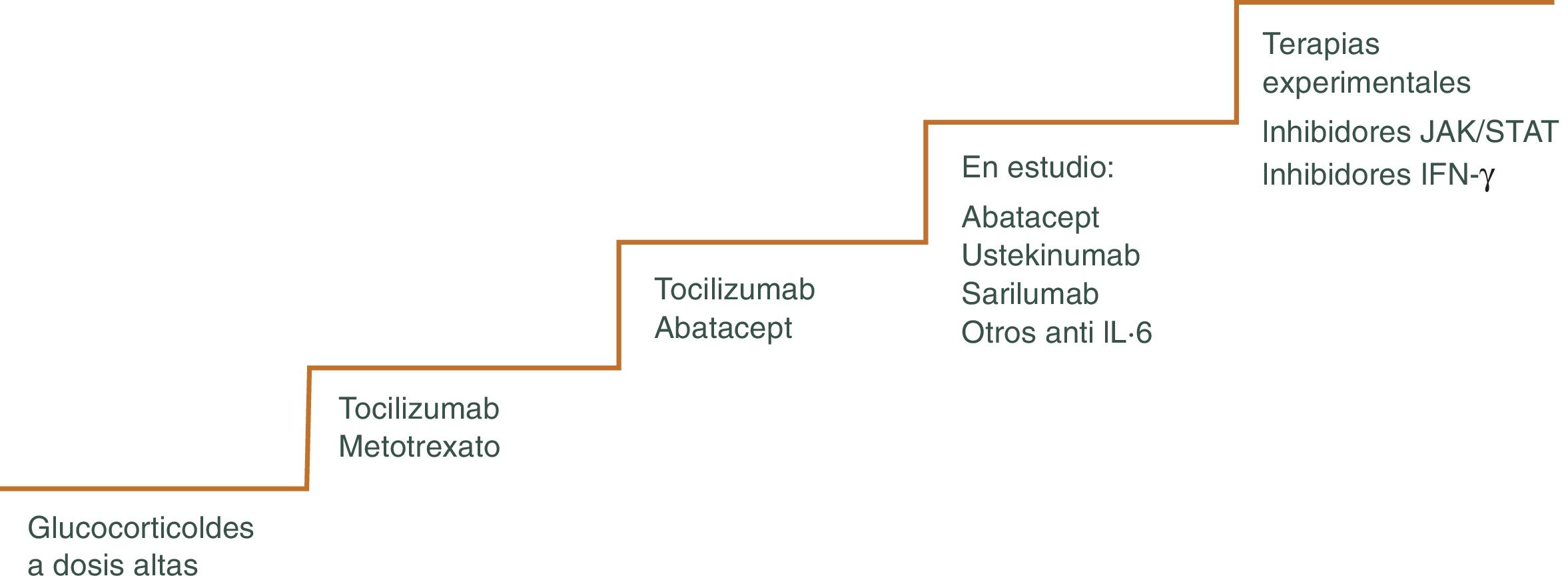
Los GC continúan siendo la piedra angular del tratamiento47,48, pero existen otros fármacos como los inmunosupresores o más recientemente terapia biológica que pueden utilizarse para minimizar los efectos adversos de los mismos, pudiendo disminuir más rápidamente los GC y el riesgo de recaídas frecuentes que ocurren a menudo cuando se reduce la dosis, sobre todo cuando se intenta bajar la dosis de prednisona por debajo de 10 mg/día49. El algoritmo terapéutico se muestra en la figura 3 y en la figura 4.
- 1.
Glucocorticoides sistémicos:


Los GC son el tratamiento de primera línea por su capacidad para reducir el riesgo de ceguera y producir un rápido alivio de síntomas y signos sistémicos. Además, si se administran a tiempo, pueden prevenir la complicación potencial más temida de la ACG, la pérdida visual permanente50–52.
La dosis de inicio depende de:
- •
Si el paciente en el momento de la sospecha clínica no presenta asociada alteración visual: administrar prednisona entre 40 a 60 mg/día, vía oral (algunos autores recomiendan una dosis inicial de prednisona de 1 mg/kg/día en dosis única, aunque algunos expertos sugieren comenzar con dosis divida en tres tomas cada ocho horas). Después de tres a cuatro semanas se realizará un descenso gradual y progresivo de dosis.
Una pauta práctica es comenzar si el paciente tiene clínica visual con prednisona a razón de 60 mg/día y si no sufre manifestaciones isquémicas graves con 40 mg/día. Después, se reducirá la dosis de forma gradual y progresiva a razón de 5 mg cada dos semanas hasta llegar a una dosis de 20 a 25 mg/día. Más tarde, se bajará la dosis a razón de 2,5 mg cada dos a tres semanas hasta llegar a 10 a 15 mg/día. Al alcanzar una dosis de 10 mg/día debemos ir reduciendo la prednisona todavía más despacio por el mayor riesgo de recaídas de la ACG, realizando una reducción aproximada de alrededor de 1,0 a 1,25 mg/día cada dos a cuatro semanas. Si el individuo experimentase recaída, volveremos a incrementar la dosis hasta aquella previa con la cual estaba libre de síntomas.
- •
Si el paciente presenta clínica visual, se remitirá a urgencias hospitalarias para iniciar tratamiento con metilprednisolona endovenosa en dosis de 500 a 1.000 mg/día durante tres días para, posteriormente, pasar a prednisona por vía oral en dosis de 60 mg/día53,54.
- 2.
Inmunosupresores ahorradores de esteroides
- 2.
Puede utilizarse en situaciones en las que se han producido o se anticipan toxicidades relacionadas con los GC. También en pacientes que sufren frecuentes recaídas al reducir la dosis de prednisona. Se considera uno de estos fármacos efectivo, si permite acortar la duración del GC y disminuir, por tanto, los efectos secundarios del mismo. Las opciones actuales más plausibles incluyen el tratamiento biológico con el inhibidor del receptor de IL-6 tocilizumab (TCZ) o con metotrexato (MTX).
MetotrexatoEl MTX es el fármaco inmunosupresor convencional más utilizado en el manejo de la ACG refractaria55–58. Sin embargo, la eficacia de este medicamento en ACG es modesta. Existen varios estudios que muestran que el MTX disminuye el riesgo de recidivas experimentada por las personas con ACG, así como su exposición total a GC. Sin embargo, no avalan su utilización sistemática ya que no hay un beneficio añadido en términos de eficacia y toxicidad. Además, se trata de un fármaco que requiere monitorización estrecha del paciente para detectar posibles efectos secundarios tóxicos.
TocilizumabEl único tratamiento aprobado actualmente en el manejo de la ACG por la Administración de Alimentos y Medicamentos de los Estados Unidos y la Agencia Europea de Medicamentos es el TCZ, antagonista del receptor de la IL-6, citocina importante en la patogénesis de la ACG. Es responsable de la síntesis de la proteína C reactiva en el hígado, activa las células B, aumentado la producción de anticuerpos y estimula la angiogénesis, produciendo un aumento de la permeabilidad vascular59. La persistencia de niveles séricos elevados de IL-6 en pacientes tratados con GC sugieren la presencia de actividad inflamatoria activa. La utilización del TCZ como ahorrador de GC se ha empleado en caso de personas con enfermedad recurrente y refractaria60. Los resultados de los ensayos clínicos muestran que tras TCZ asociado a un descenso gradual de GC durante un año, los pacientes permanecen en remisión y se requieren dosis inferiores de GC en comparación con su uso aislado61–64.
El tratamiento con TCZ tiene efectos secundarios, siendo el más frecuente las infecciones, seguido de reacciones de hipersensibilidad, hemorragia, perforación gastrointestinal, cánceres, etc. Su uso concomitante con un corticoide aumenta el riesgo de inmunosupresión y, por tanto, de infecciones.
Cabe destacar que este fármaco comercializado en España ha sufrido modificaciones con respecto a su inclusión en la prestación farmacéutica del Sistema Nacional de Salud Español, incluyendo la imagen como criterio diagnóstico65–67.
Otras terapias biológicasEn la ACG, los antagonistas del TNF- α son ineficaces o solo parcialmente eficaces68,69.
Existen otros tratamientos que se han propuesto como terapia ahorradora de GC, pero su uso no puede respaldarse debido a su eficacia inferior, toxicidad potencial y falta de estudios. Entre ellos destacan: Abatacept vía subcutánea (SC) o intravenosa (IV)70, azatioprina, vía oral (VO) o IV71, ustekinumab IV o SC72, ciclofosfamida, VO o IV73. Con estas terapias la experiencia es escasa y por ahora su eficacia no ha sido refrendada en grandes estudios.
- 3.
Otros tratamientos: antiplaquetarios/anticoagulantes
Un metaanálisis74 valoró el efecto de la terapia antiplaquetaria/anticoagulante establecida sobre la aparición de complicaciones isquémicas graves en pacientes con ACG en su evolución y tratamiento con GC. Teniendo en cuenta estos datos, el uso de aspirina podría tener un cierto beneficio marginal para evitar eventos isquémicos una vez realizado el diagnóstico de la ACG sin riesgo hemorrágico asociado.
RecomendacionesEl médico de Atención Primaria debe participar de manera conjunta con el servicio de reumatología en la evolución clínica de la ACG, los objetivos del seguimiento son:
- -
Detectar precozmente las recaídas,
- -
controlar la seguridad farmacológica y
- -
evidenciar secuelas isquémicas
Se recomienda, por tanto, realizar:
- A)
Seguimiento:
- •
Mensual durante los primeros seis meses de tratamiento.
- •
En cada visita, a los pacientes se debe realizar una anamnesis rutinaria sobre los síntomas craneales, así como de cualquier síntoma nuevo o empeoramiento de los mismos, individualizando los intervalos de citas, analíticas y descenso de dosis de GC.
- •
- B)
Prevención de efectos secundarios del tratamiento:
- -
Glucocorticoides: vigilar la osteoporosis/gastropatías inducida por GC, recomendando tratamiento con inhibidores de la bomba de protones o con antagonistas H2, según valoración de riesgo de sangrado hasta que se alcancen dosis de corticoides de 10 mg/día así como suplementos de calcio/vitamina D y bifosfonatos, según tiempo y valoración de riesgo de fracturas, aparición o empeoramiento de diabetes tipo 2, hipertensión arterial, cataratas, glaucoma, atrofia muscular y el aumento del riesgo de infecciones oportunistas75,76.
- -
Metotrexato: se debe controlar la toma de ácido fólico de forma coadyuvante, monitorizar con analítica mensual la función renal, enzimas hepáticas y hemograma77.
- -
Tocilizumab: vigilar la aparición de infecciones, insuficiencia cardiaca y citopenia grave. Es recomendable el control analítico con hemograma y bioquímica (incluyendo perfil hepático) mensual durante los primeros tres meses78.
- -
Este trabajo no ha recibido ningún tipo de financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Aguado Castaño, Ana Carlota. Centro de Salud Babel, Alicante.
Aicart Bort, María Dolores. Centro de Salud Rafalafena, Castellón.
Babiano Fernández, Miguel Ángel. Centro de Salud Almadén, Ciudad Real.
Caballer Rodilla, Julia. Centro de Salud El Restón, Madrid.
Cabrera Ferriols, María Ángeles. Centro de Salud San Vicente del Raspeig, Alicante.
Carrasco Carrasco, Eduardo. Centro de Salud de Abarán, Murcia.
Frías Vargas, Manuel. Centro de Salud Comillas, Madrid.
Fuertes Domínguez, Diana. Centro de Salud Cervera de Pisuerga, Palencia.
García Fernández, Pedro. Centro de Salud Nueva Málaga, Málaga.
García Lerín, Aurora. Centro de Salud Almendrales, Madrid.
Gil Gil, Inés. Centro de Salud Vielha, Lleida.
García Vallejo, Olga. Centro de Salud Comillas, Madrid.
López Téllez, Antonio. Centro de Salud Puerta Blanca, Málaga.
Mayorga Criado, Alberto. Centro de Salud Llefia, Badalona
Peiró Morant, Juan. Centro de Salud Ponent, Islas Baleares.
Perdomo García, Frank J. Urgencias, Hospital La Paz, Madrid.
Piera Carbonell, Ana. Centro de Salud Luanco-Gozón, SESPA, Asturias.
Pietrosanto, Teresa. Centro de Salud San Vicente del Raspeig, Alicante.
Ramírez Torres, José Manuel. Centro de Salud Puerta Blanca, Málaga.
Robledo Orduña, Carlos. Centro de Salud San Vicente del Raspeig I, Alicante.
Vázquez Gómez, Natividad. Centro de Salud Auxiliar Moncófar, Castellón.
Este documento ha sido validado por la Comisión Nacional de Validación de la Sociedad Española de Atención Primaria (SEMERGEN) con el aval no. 00194-2020.
